2019/06/28 産業技術総合研究所
発表のポイント
- 計算機科学により、膨大な数のペプチド注1群から標的タンパク質の天然変性領域に結合する医薬品候補ペプチドを推定し、合理的で迅速な創薬研究を可能に
- 本手法を用い、8×10の16乗通り(8億×1億通り)のペプチド群から、天然変性領域を持つがん抑制タンパク質p53の機能を制御する人工ペプチドを発見
概要
東北大学多元物質科学研究所の鎌形清人准教授、産業技術総合研究所人工知能研究センターの亀田倫史主任研究員、および立命館大学薬学部創薬科学科の北原亮教授らの研究グループは、タンパク質の天然変性領域を標的とした医薬品候補ペプチドの設計法の開発に成功しました。
医薬品の開発には、疾患に関与するタンパク質が持つ特定の立体構造に結合し、そのタンパク質の機能を、阻害または促進する低分子の設計が行われています。しかし、特定の立体構造を持たない天然変性領域を持つタンパク質に対しては上述の方法が通用せず、医薬品の迅速な開発は困難でした。研究グループは、この天然変性領域のアミノ酸配列情報のみを使用し、計算機内で、この領域に特異的に結合する医薬品候補ペプチドを迅速に設計する方法を開発しました。本手法を用いて、8×10の16乗通り(8億×1億通り)のペプチド群から、天然変性領域を持つがん抑制タンパク質p53の機能を制御する人工ペプチドを発見しました。この人工ペプチドは医薬品候補分子として期待されます。以上より、本手法は、タンパク質の天然変性領域を標的とする創薬研究を加速する可能性があります。
本研究成果は、2019年6月28日(英国夏時間)に英国科学誌Scientific Reports(オンライン版)に掲載されました。また、本研究は、科学研究費助成事業、および産総研-東北大マッチング事業の支援を受けて、実施されました。
研究背景
一般に、医薬品は疾患に関与するタンパク質の特定の立体構造に結合し、その機能を阻害、または、促進します。また、コンピューター上でタンパク質の立体構造に対して、様々な化合物を結合させ医薬品候補を絞り込むことで、医薬品開発を効率的、合理的に行うことが可能になりました(Fig. 1a)。しかし、特定の立体構造を持たない領域(天然変性領域)を含むタンパク質が、全タンパク質の約30%を占めること、また、この天然変性領域がアルツハイマー病やがんなどの疾患に関わることが明らかになってきました。特定の構造を持たない天然変性領域は、絶えず構造が変化するため、特定の構造としか結合できない医薬品候補の化合物とは強く結合できません(Fig. 1b)。そのため、上記の手法ではタンパク質の天然変性領域をターゲットとした医薬品の開発は困難でした。

Fig.1 医薬品候補分子の探索。 a)タンパク質の立体構造に対して様々な分子をドッキングさせ、形状が合致した分子を医薬品候補とします。 b)タンパク質が特定の形を持たない場合(天然変性)、ドッキング計算による探索ができません。
研究の成果
研究グループは、特定の立体構造をとらない天然変性領域に柔軟に結合する“ペプチド”と、タンパク質の立体構造を安定化する“アミノ酸ペアの結合力”に着目し、天然変性領域をターゲットとした創薬法を開発しました(Fig. 2)。ペプチドを設計する場合1残基あたり20種のアミノ酸候補があり、候補ペプチドの数は膨大な数にのぼるため、全ての候補ペプチドを作成し実験的に検証することは困難でした。本手法では、計算機の中で、MiyazawaとJerniganが開発したアミノ酸ペア結合エネルギー(MJエネルギー)注2を用いて、ターゲットタンパク質の天然変性領域に結合する候補ペプチドを迅速に絞り込むことができます(Fig. 2)。MJエネルギーは、アミノ酸ペアを形成しやすい・しにくい傾向を表現している指標として考えることができるので、ペプチド全体で、MJエネルギーができるだけ大きくなるような配列を選ぶことで、強く結合するペプチドを設計できるのではないかと我々は考えました。
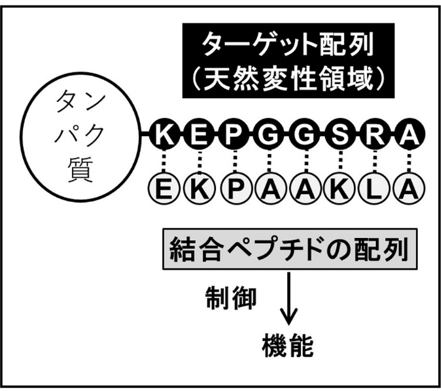
Fig.2 設計ペプチドによる天然変性タンパク質の機能制御。標的である天然変性領域のアミノ酸配列に対して、MiyazawaとJerniganによるアミノ酸ペア結合エネルギーが高いアミノ酸を並べ設計ペプチド(結合ペプチド)とします。設計ペプチドがタンパク質の天然変性領域に結合することで、タンパク質の機能の制御を可能にします。
例えば、アミノ酸は20種類からなるため、2アミノ酸から構成されるペプチド全種類の数は20×20=400通りあります。しかし、13アミノ酸から構成されるペプチドの場合、そのアミノ酸配列の数は、20×20×・・・×20=2013≒8.2×1016=約8垓2000京通り(およそ8億×1億通り)という天文学的な数となってしまいます(Fig. 3a)。我々の手法では、ターゲットとなるアミノ酸に対して、最も大きいMJエネルギーを持つアミノ酸を選ぶことで設計するので、迅速に候補配列を選ぶことができます(Fig. 3b)。
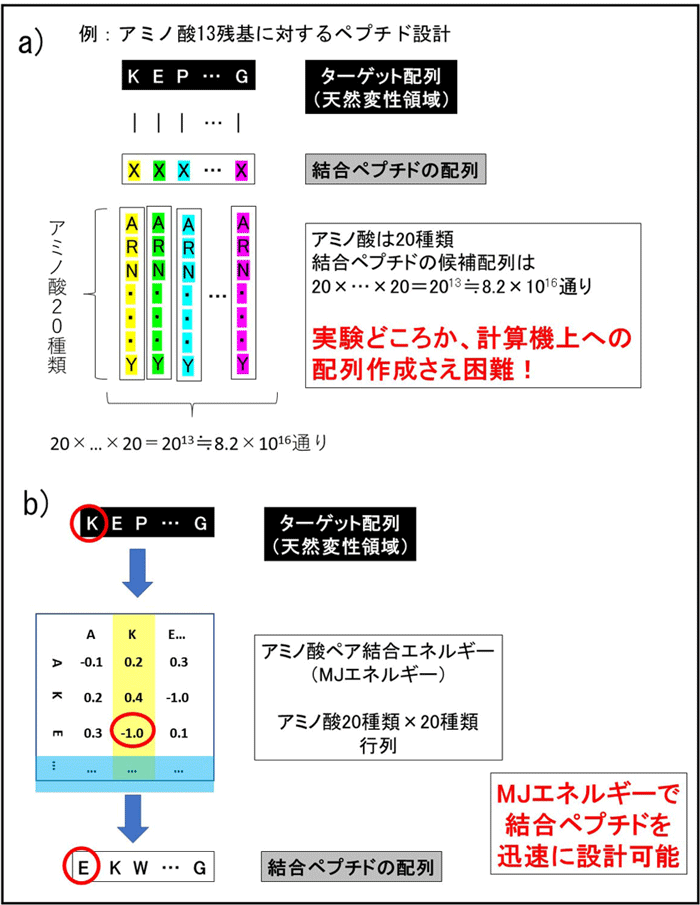
Fig.3 ペプチドの設計法 a) 天然変性タンパク質のターゲット配列に結合するペプチドの候補の数。 b) MiyazawaとJerniganによるアミノ酸ペア結合エネルギー(MJエネルギー)を用いた結合ペプチドの迅速な設計。
本手法を検証するため、モデルとしてがん抑制タンパク質p53の天然変性領域を対象とした研究を行いました。まず、p53の天然変性領域に強く結合する13残基、および16残基の候補ペプチドを作成することにしました。次に、絞り込んだ候補ペプチドを合成し、滴定実験、NMR、および分子動力学シミュレーションを行ったところ、候補ペプチドの1つがp53の天然変性領域に選択的に結合し、強い結合力(~μMの解離定数)を持つことが分かりました(Fig. 4a,b)。さらに、滴定実験と単分子蛍光計測を行ったところ、この設計ペプチドがp53のDNAへの結合を阻害し、DNA上で滑る動きを抑制することを明らかにしました(Fig. 4c)。以上より、開発した手法を用いて、p53の天然変性領域に結合し、その機能を制御できるペプチドを合理的に設計できることを実証しました。
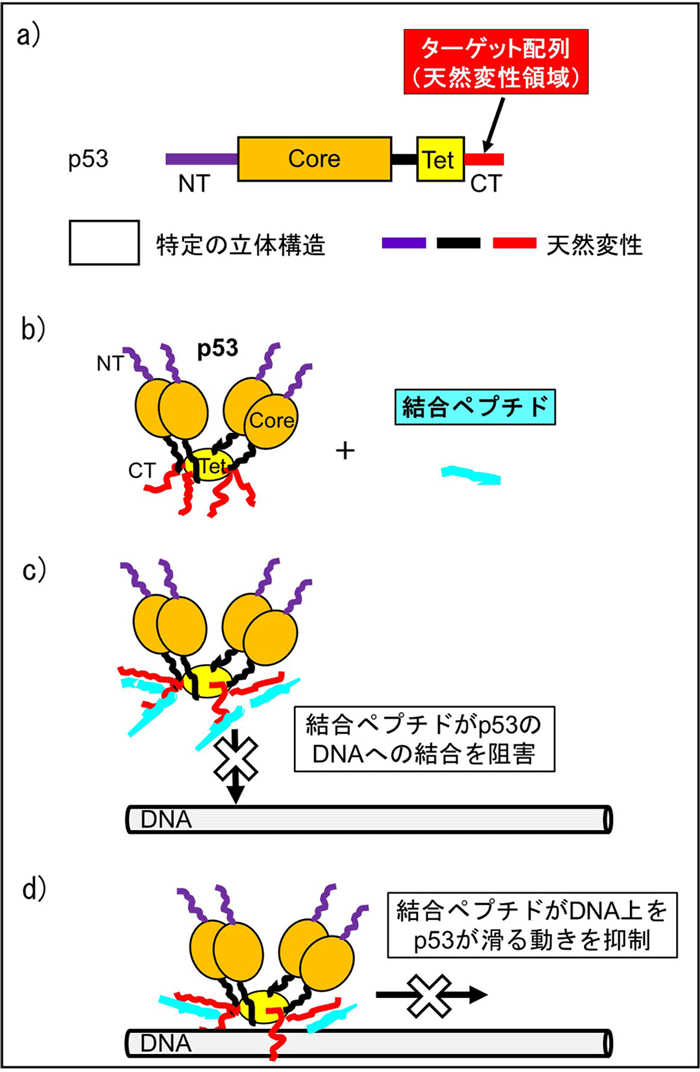
Fig.4 結合ペプチドによるp53の機能の制御。a) p53の一次構造(アミノ酸配列)と立体構造の関係。NT、core、Tet、CTは、それぞれ、N末ドメイン、コアドメイン、4量体ドメイン、C末ドメインを表しています。CTをターゲット配列として、結合ペプチドの設計を行いました。b) p53の3次元立体構造と結合ペプチド。c) 結合ペプチドによるp53のDNAへの結合の阻害。d) 結合ペプチドによるDNA上でのp53の動きの抑制。
本手法は、通常では設計が難しい、タンパク質の天然変性領域を標的とする医薬品候補ペプチドを容易に設計できることから、創薬研究を加速することが期待されます。
論文情報
題目:Rational design using sequence information only produces a peptide that binds to the intrinsically disordered region of p53
著者:Kiyoto Kamagata1*, Eriko Mano1, Yuji Itoh1,2, Takuro Wakamoto3, Ryo Kitahara4, Saori Kanbayashi1, Hiroto Takahashi1, Agato Murata1,2, and Tomoshi Kameda5*
所属:1東北大学 多元物質科学研究所, 2東北大学大学院 理学研究科化学専攻, 3立命館大学大学院 生命科学研究科, 4立命館大学 薬学部創薬科学科, 5産業技術総合研究所 人工知能研究センター
雑誌:Scientific Reports
URL:www.nature.com/articles/s41598-019-44688-0
DOI:10.1038/s41598-019-44688-0
用語解説
- 注1)ペプチド
- アミノ酸が連結した高分子鎖。
- 注2)Miyazawa-Jernigan(MJ)エネルギー:
- 立体構造を分かっているタンパク質を多数集めてきて、それらの中から、互いに近距離にあるアミノ酸ペアの個数を数え上げ、その頻度をエネルギーとして表現したもの。立体構造群の中でのアミノ酸ペアの数が多ければ、そのアミノ酸ペアのエネルギーの値も大きくなります。つまり、MJエネルギーはアミノ酸ペアを形成しやすい・しにくい傾向を表現している指標ということが言えます。

