2019-08-20 東京大学
松井 求(生物科学専攻 助教)
岩崎 渉(生物科学専攻 准教授)
発表のポイント
- ネットワーク解析技術を分子系統学へ応用することで、「グラフスプリッティング法(GS法)」という新しい系統解析手法を開発し、その有効性を示した。
- GS法を用いることで、これまで解く事が難しかった非常に長い進化時間を経た遺伝子群の進化過程を初めて明らかにした。
- GS法は、幅広い問題に適用可能な強力な手法であり、同時に既存の系統解析手法や多重配列アライメントの性能向上にも寄与することから、様々な波及効果が期待される。
発表概要
「原初の生命はどのようなものであったのか?」「そこから現在に至るまで生命はどのように進化してきたのか?」これらはどれも進化学者が長年取り組み続けてきた「究極の問い」です。これらの問いに答えるためには、細菌・古細菌・真核生物にわたる多様な生物の遺伝子データをもとに、極めて長い進化史を分子系統解析(注1)によって解き明かすことが必要不可欠です。しかし、この規模の進化史を解く事は、既存の分子系統解析手法(注2)にとってはしばしば困難でした。その原因は、既存手法のいずれもが出発点としている多重配列アライメント(Multiple sequence alignment; MSA、注3)の精度がこうした場合には著しく低下してしまうことにあります。そこで我々は、MSAに代えて配列類似性グラフ(Sequence similarity graph; SSG、注4)を出発点とする新たな分子系統解析手法「グラフスプリッティング法(GS法)」を新たに開発しました。進化シミュレーションに基づく検証により、GS法は既存手法よりも高い精度で系統樹を推定可能であることが示されました。さらに、実際の遺伝子配列データに基づく検証の結果、多くの遺伝子群についてGS法は既存手法よりも信頼性の高い系統樹を推定できることが分かりました。特に、最大の遺伝子群であるTIM-barrelスーパーファミリー(注5)への適用結果から、生命の初期進化にまつわる有力な仮説「RNAワールド仮説」(注6)を支持する結果が得られました。今後、分子系統解析のみならず進化学の幅広い領域におけるGS法の貢献が期待されます。本研究は、現地時間2019年8月22日(木)付「Systematic Biology」誌に掲載されます。
発表内容
「最初の生命はどのようなものであったのか?」「そこから我々人類に至るまで生命はどのように進化してきたのか?」といった問題に答えるためには、現在の地球上に生きる様々な生命の共通の祖先がどのような遺伝子を持っていたのか、また、そうした遺伝子がはるかな時間を経てどのように進化してきたのかを明らかにする必要があります。しかし、既存の系統解析手法(以下、既存手法)でこうした問題を解く事は困難でした。その理由の一つは、遺伝子群が経た進化時間が長ければ長いほど、その「多重配列アライメント(Multiple sequence alignment; MSA)」の精度が著しく低下してしまうことにあります。既存手法はMSAの情報を元に系統樹を推定するため、MSAが不正確な場合、推定される系統樹も不正確になってしまうのです(図1)。

図1:多重配列アライメントと配列類似性グラフの比較
進化的に遠く離れている場合、遺伝子PとQ、およびQとRは整列可能だが、PとRは整列不可能である、というケースがでてくる。多重配列アラインメントではこのような場合、三つの遺伝子で共有される縦の列がうまく取得できず(左)、多重配列アライメントから構築する距離行列にも「無限大」という扱いの困難な距離が出現する(中央)。一方で配列類似性グラフでは、PとRが整列できないことをPとRの間に「エッジを張らない」ということで自然に表現することができる(右)。GS法は、この利点を最大限生かすことでこれまでの限界を乗り越える方法である。NJ=Neighbor Joining(近隣結合法)、MP=Maximum Parsimony(最節約法)、ML=Maximum Likelihood(最尤法)、BI=Bayesian Inference(ベイズ法)。
例えば、遥か昔に存在していた遺伝子を共通祖先として持つ遺伝子群(タンパク質スーパーファミリー)に関して、現在知られている遺伝子群の大多数についてはMSAを構築することが実際に困難であることが報告されています。
そこで我々は、出発点をMSAではなく配列類似性グラフ(Sequence similarity graph; SSG)とする「グラフスプリッティング法(GS法)」という新たな系統解析手法を考案しました(図2)。
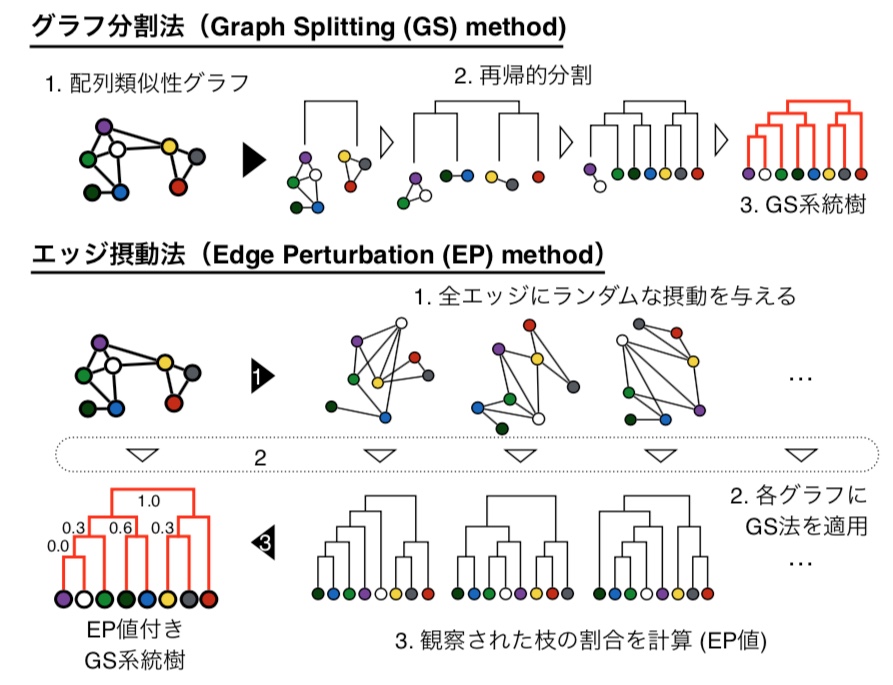
図2:グラフスプリッティング(GS)法とエッジ摂動(EP)法の概念
(上)GS法では、まず初めに配列類似性グラフを構築する。次にそのグラフを再帰的に二分割していく。ここでスペクトラルクラスタリングという方法を使ってグラフを分割することで、高速かつ正確な分割を実現している。全ての要素がバラバラになるまで再帰的分割を行ったのち振り返ると、その分割過程がGS系統樹となっている。 (下)EP法では、配列類似性グラフのエッジにランダムな摂動を与えることでエッジの長さを変えたグラフを大量に作成する。次に各グラフに対してGS法を適用して系統樹を構築し、観察された内部枝の頻度(EP値)を計算する。最後に摂動を与えていないオリジナルの配列類似性グラフを元に構築したGS系統樹の各内部枝にEP値を付与する。EP値が高いほど(1.0に近いほど)再現性の高い、つまり信頼できる内部枝であることを意味する。
GS法は、まずMSAの代わりにSSGを構築し、次にSSGを再帰的に分割することで系統樹を推定します。このようにしてMSAに由来する問題を丸ごと回避します。また、GS系統樹の内部枝を評価する方法「エッジ摂動法(Edge Perturbation method; EP法)」も合わせて開発しました(図2)。EP法は、まず配列類似性グラフのエッジにランダムな摂動を与えることでエッジの長さを変えたグラフを大量に作成し、次に各グラフに対してGS法を適用して系統樹を推定し、最後に観察された内部枝の頻度(EP値)を計算します。EP値が高いほど再現性の高い、つまり信頼できる内部枝であることを意味します。
大規模シミュレーションに基づく性能検証の結果、特に互いに進化的に遠く離れている遺伝子群について、GS法は既存手法よりも良い精度で系統樹を推定できること、系統解析手法における問題としてよく知られている長枝誘引などの問題に対して頑健性をそなえていること、計算速度がこれまで最速の手法であった近隣結合法と同程度以上であることが示されました(図3、4)。
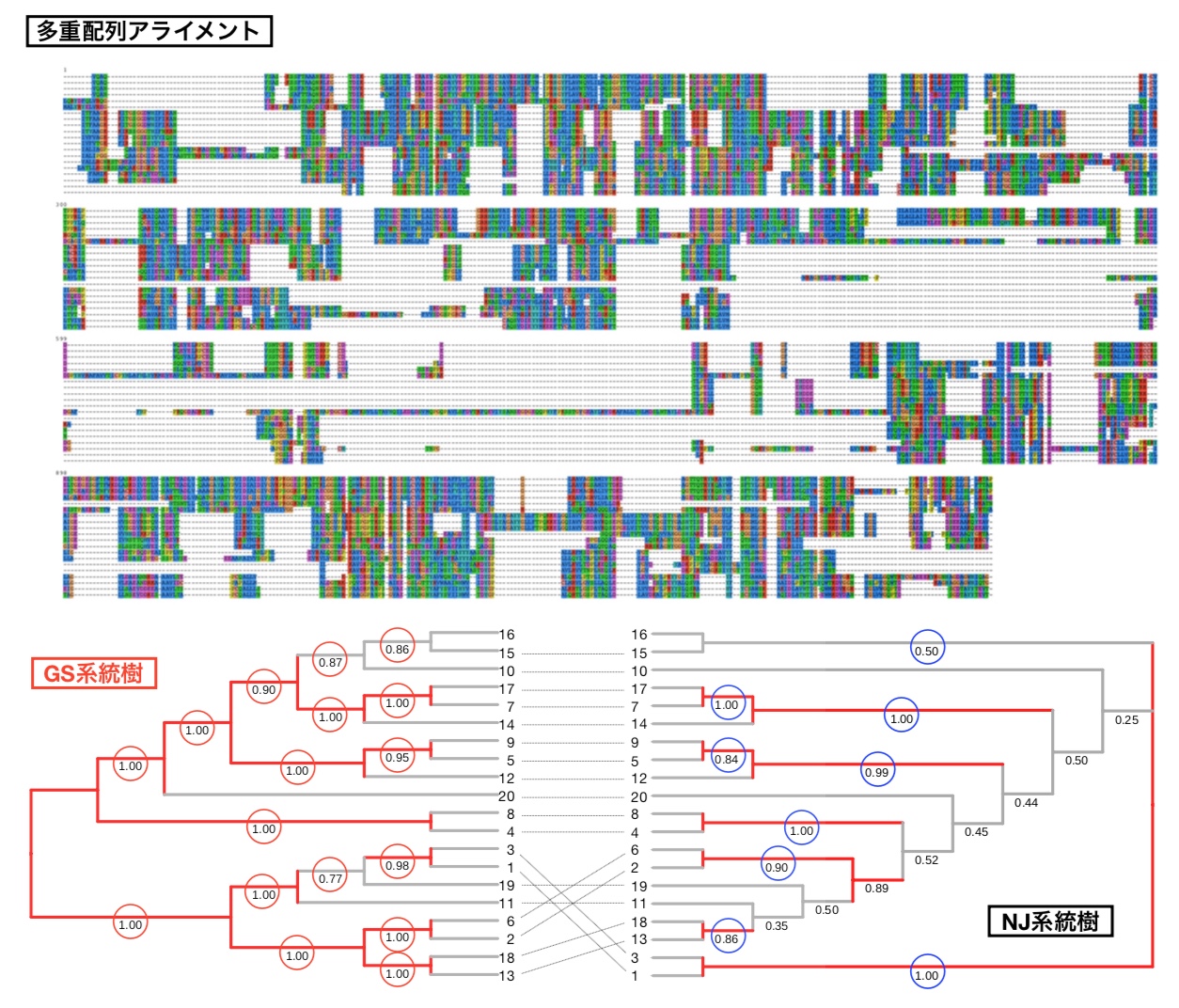
図3:多重配列アライメントと系統樹の例(進化的に遠く離れている場合)
(上)進化シミュレーションによって作成した20本の配列を整列した例。文字はアミノ酸、ハイフンはギャップを表している。進化的に遠く離れているため縦一列で揃っている場所が少ない。このようなケースでは、多重配列アライメントに基づく系統解析は困難である。(下)GS系統樹とNJ系統樹(NJ=近隣結合法、既存系統解析手法の一つ)の比較。丸は正しい枝、数字は枝の信頼度を示す。GS系統樹は全ての枝が正しかった一方で、NJ系統樹はほぼ半分の枝が間違っていた。
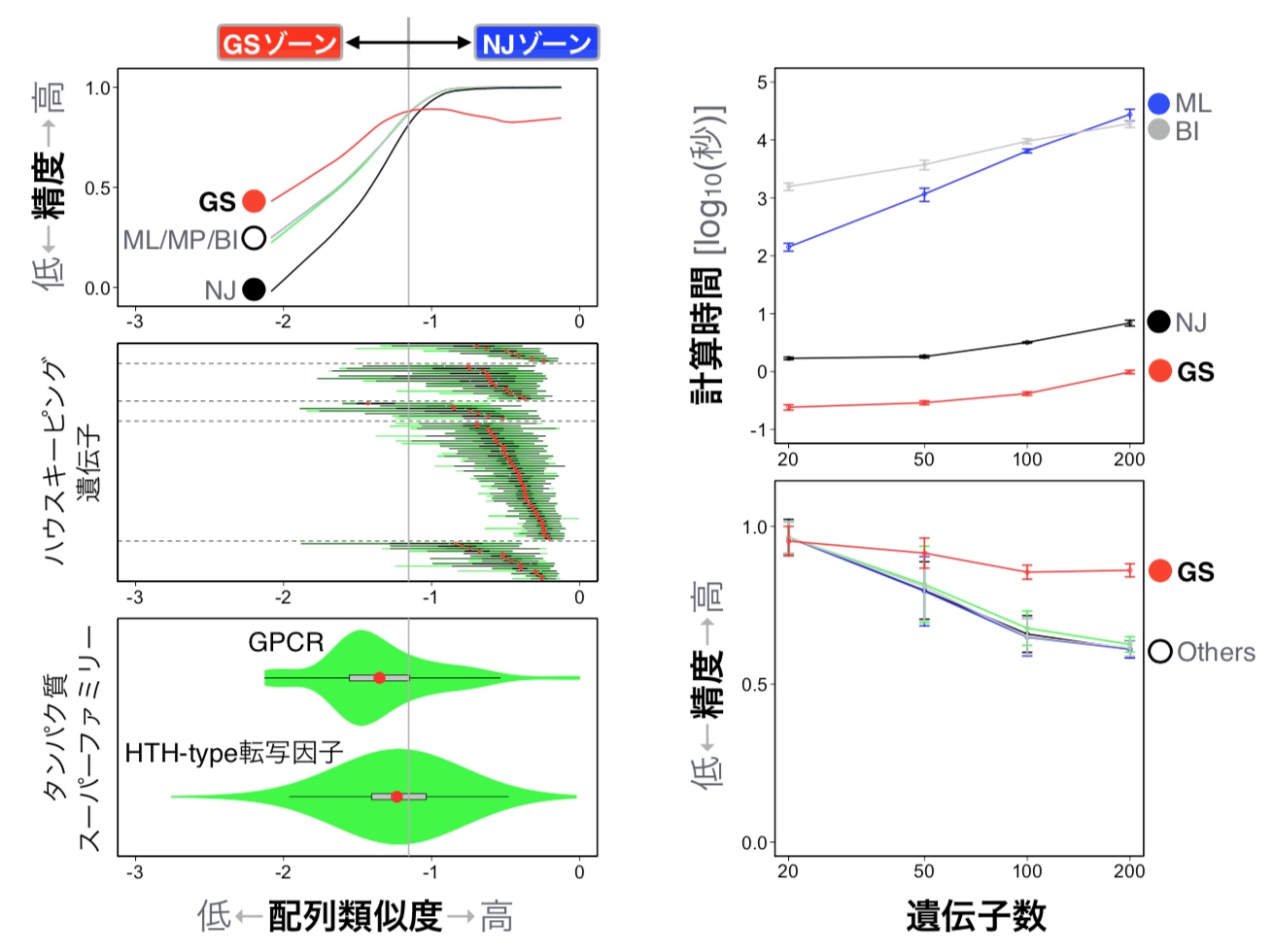
図4:GS法と既存手法の精度および実行時間の比較
(左上)各系統解析手法で構築した分子系統樹の精度比較。配列類似度が低い場合(GSゾーン)、GS法はどの既存手法よりも良い精度を示した。一方で配列類似度が高い場合(NJゾーン)では、既存手法はほぼ同じ精度を示し、GS法を上回った。(左中)ハウスキーピング遺伝子の配列類似度の分布。NJゾーンに集中していることが分かる。(左下)タンパク質スーパーファミリーの配列類似度の分布。GSゾーンに分布していることから、ハウスキーピング遺伝子のような互いの遺伝子距離が比較的小さな遺伝子群については既存手法を用いるのが適切であり、一方で、タンパク質スーパーファミリーのような遺伝子距離が大きな問題についてはGS法を用いることが適切である、ということになる。すなわち、配列類似度にしたがって既存手法とGS法を使い分けることがより良い系統樹の推定方法であることが分かった。(右上)計算時間の比較。GS法はこれまで最も速い手法の一つであったNJ法を上回る計算速度を示した。(右下)解析する遺伝子数を増やした場合の精度の比較。一般に解析する遺伝子数が増えると分子系統解析の精度が落ちることが知られているが、GS法は遺伝子数の増加に対して頑健性を示した。NJ=Neighbor Joining(近隣結合法)、MP=Maximum Parsimony(最節約法)、ML=Maximum Likelihood(最尤法)、BI=Bayesian Inference(ベイズ法)。
また、実データへの適用例として、これまで知られているなかで最大の遺伝子群であるTIM-barrelスーパーファミリーにGS法を適用したところ、ピリミジン代謝の初期進化過程の一端を明らかにすることができ(図5、6)、それは初期進化にまつわる有力な仮説「RNAワールド仮説」を支持するものでした。さらに、データベースに登録されている243のタンパク質スーパーファミリーそれぞれに対してGS法を適用した結果、多くのスーパーファミリーについて信頼性の高い系統樹を推定することができました。
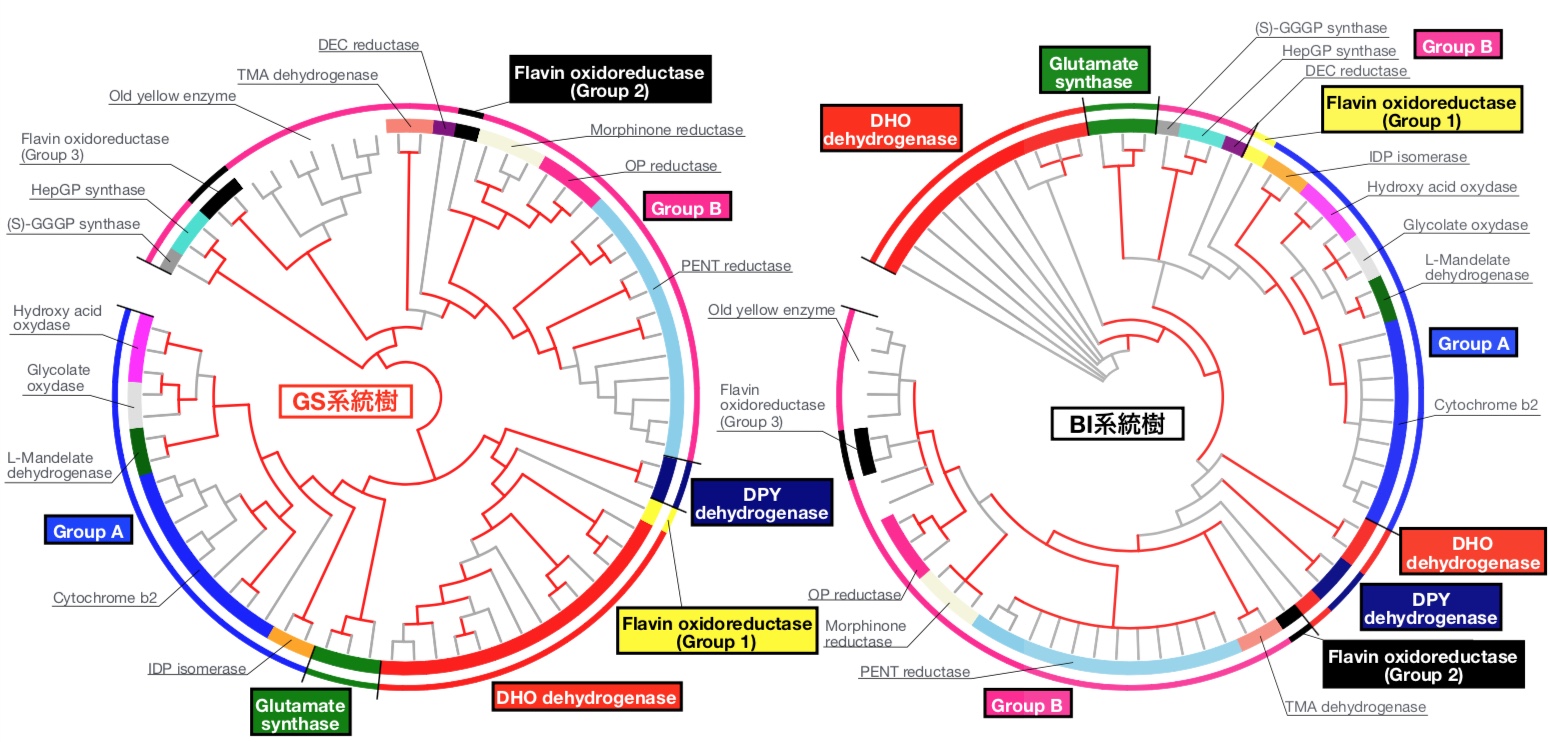
図5:TIM-barrelスーパーファミリーの分子系統樹
GS法によって推定した系統樹(左)とBI(ベイズ)法によって推定した系統樹(右)の比較。赤い枝(グレートーンで印刷された場合:より黒に近いグレーの枝)は高い支持率を持つ内部枝であることを示し、外円の色は異なる色ごとに異なる遺伝子の機能を表している。GS系統樹の方がBI系統樹よりも支持された内部枝の数が多く、全体的に信頼性の高い系統樹であることが分かる。また、ピリミジン合成に関わるDihydroorotate (DHO) dehydrogenaseとDihydropyrimidine (DPY) dehydrogenaseの遺伝子がそれぞれ単系統としてまとまり、また互いに進化系統樹上で近縁であると推定されたことは、これらの酵素が生命の進化史において非常に早い段階で誕生したとする「RNAワールド仮説」と整合的な結果である。
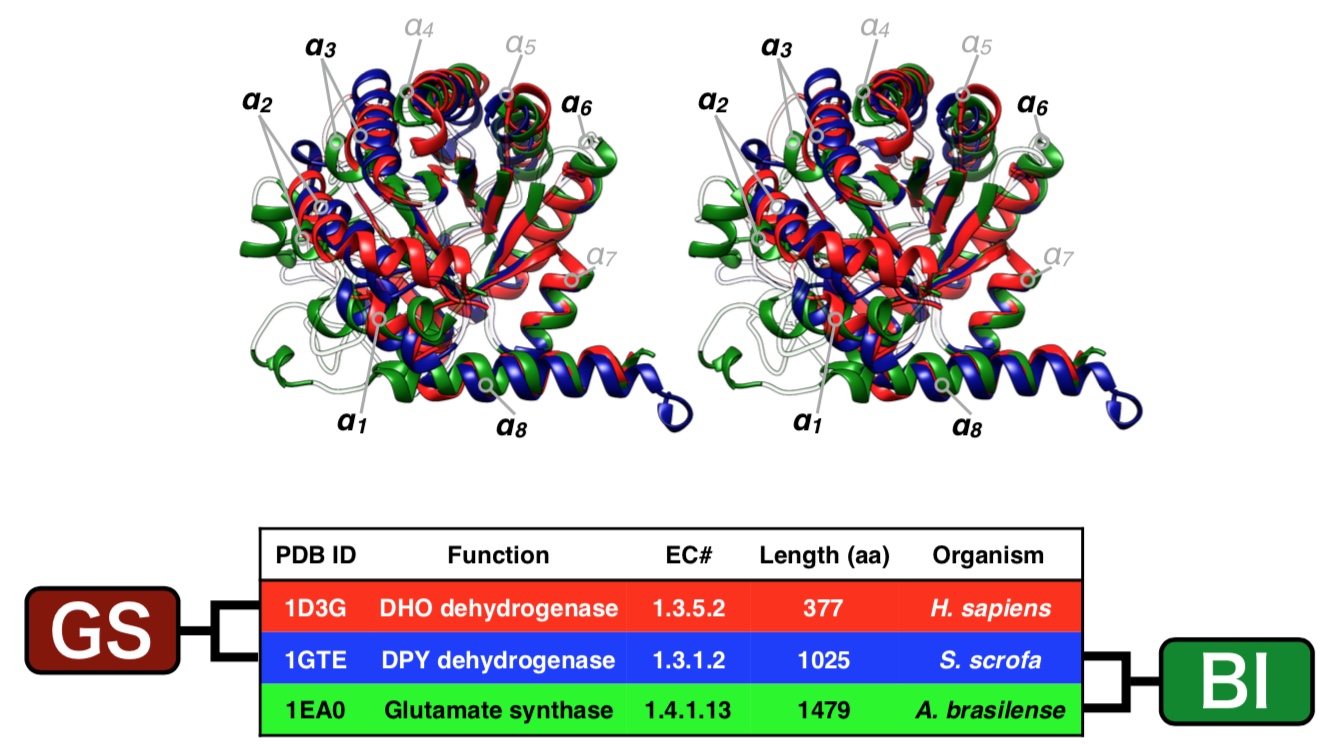
図6:TIM-barrelスーパーファミリーに属するタンパク質の立体構造比較
図5の系統樹の確からしさをタンパク質の立体構造の比較に基づいて検証した。この検証にあたっては「進化的により近ければタンパク質の立体構造もより似ているだろう」という仮定を置いている。(上)DHO dehydrogenase(赤)、DPY dehydrogenase(青)、およびGlutamate synthase(緑)を重ねたステレオグラム。立体構造の色と下表の色は一致している。特に黒文字(α1, α2, α3, α6, α8)で示した部分構造(αヘリックス)については、DHO dehydrogenaseとDPY dehydrogenaseの立体構造の方がGlutamate synthaseよりも近いことが分かる。(下)GS法ではDHO dehydrogenaseとDPY dehydrogenaseがより進化的に近いと推定した一方で、BI法はDPY dehydrogenaseとGlutamate synthaseの方がより進化的に近いと推定した(図5参照)。つまり、GS系統樹の確からしさは立体構造解析からも支持されたことになる。
特にDNA/RNAポリメラーゼやT-foldをはじめとするスーパーファミリーは系統樹全体にわたって内部枝が強く支持されており、代謝、転写・複製、膜輸送の初期進化を照らす新たな知見が得られました。
さらに、GS法で推定した系統樹をガイドツリーとして使用すると、既存手法の弱点となっていたMSAの精度が向上することも分かりました。このことはGS法が、MSAの精度向上を介して、既存の系統解析手法の性能をも向上させることが可能であることを示唆しています。今後、進化学における幅広い問題において、GS法が大いに貢献すると期待されます。
本研究は、文部科学省科学研究費補助金、日本学術振興会科学研究費補助金、キヤノン財団の支援を受けて実施されました。また大規模計算は国立遺伝学研究所、および京都大学化学研究所のスーパーコンピュータシステムを使用して行いました。
発表雑誌
- 雑誌名
Systematic Biology論文タイトル
Graph Splitting: A Graph-Based Approach for Superfamily-scale Phylogenetic Tree Reconstruction著者
Motomu Matsui, and Wataru IwasakiDOI番号
10.1093/sysbio/syz049論文URL
Just a moment...doi.org

用語解説
注1 分子系統解析
生物や遺伝子は、進化の過程で共通の祖先から枝分かれを繰り返すことで多様性を増してきた。この枝分かれの様子を樹木になぞらえて表現したものを系統樹と呼ぶ。近年では、DNAなどの分子レベルの情報を用いた統一的・客観的な手法で系統樹を推定することが広く行われるようになり、こうした解析を分子系統解析と呼ぶ。
注2 分子系統解析手法
分子系統樹を構築するために使われる現在の主な手法は、最節約法(Camim JH and Sokal RR, 1965)、最尤法(Felsenstein J, 1981)、近隣結合法(Saitou N and Nei M, 1987)、ベイズ法(Yang ZH and Rannala B, 1997)である。これらはいずれも後述する多重配列アライメント(MSA)を出発点にした方法で、MSAが良いアライメントである限り良い性能を示す。しかし、タンパク質スーパーファミリーのように、長い時間をかけて進化してきた結果、互いに配列の類似性が非常に小さくなってしまった遺伝子群に対しては、MSAの精度が著しく減少してしまうためにいずれの既存手法も適用困難だった。
注3 多重配列アライメント(Multiple sequence alignment; MSA)
複数の配列が与えられたとき、それぞれの配列で対応するアミノ酸や塩基を縦一列に整列すること、あるいは整列したものを多重配列アライメント(MSA)と呼ぶ。既存の分子系統解析手法はいずれもMSAに依存した方法である。
注4 配列類似性グラフ(Sequence similarity graph; SSG)
遺伝子を頂点とし、遺伝子間の配列類似性をエッジとしたネットワークを配列類似性グラフ(SSG)と呼ぶ。
注5 TIM-barrelスーパーファミリー
最大のタンパク質スーパーファミリー。樽型の特徴的な構造(TIM-barrel構造)を持つという共通点があり、様々な合成酵素からシトクロームにいたるまで多様かつ重要なタンパク質を含んでいる。立体構造が高度に保存されていることから同じ祖先遺伝子から進化してきたと考えられている一方で、遺伝子配列は互いに大きく異なっており、本スーパーファミリー全体の分子系統樹を構築することはこれまでできていなかった。
注6 RNAワールド仮説
生命の起源を説明する有力な仮説の一つ。生命は、遺伝的機能と触媒機能を併せ持つRNAが自己複製することによって形成された世界「RNAワールド」から生まれたとする。GS法を用いたTIM-barrelスーパーファミリーの分子系統解析の結果、ピリミジン合成酵素が他の酵素に先駆けて存在していたことが示唆された。これはRNAワールドが先にあり、その後RNAとタンパク質が共存する世界になったとする本仮説と整合的な結果である。
―東京大学大学院理学系研究科・理学部 広報室―

