2020-01-20 理化学研究所
理化学研究所(理研)生命機能科学研究センターバイオインフォマティクス研究開発チームの露崎弘毅特別研究員、二階堂愛チームリーダーらの共同研究チームは、大規模1細胞発現データを高精度・高速・低メモリで主成分分析(PCA)[1]する手法の性能評価を行いました。
本研究成果は、大規模な遺伝子発現データからの疾患関連細胞や遺伝子の発見で利用されるアルゴリズムの高速化・軽量化に貢献すると期待できます。
近年、臓器が持つ全細胞種を1細胞RNAシーケンス法(1細胞RNA-seq)[2]で同定する研究が盛んです。この方法で得られたデータをPCAで簡素化し、細胞の種類や数、機能を特定しますが、大規模研究では細胞の数が100万を超えるため、従来法ではそもそも計算できなかったり、膨大な計算時間、メモリ量が必要とされます。
今回、共同研究チームは、10種のPCAアルゴリズムを比較しました。その結果、高速化や低メモリ化には、行列の非ゼロ要素のみを格納する疎行列フォーマット[3]の利用や、行列の一部を逐次的に計算に用いるOut-of-core[4]な実装が有効なことが分かりました。そして、それらを考慮したソフトウェアを実装し、その有効性を示しました。
本研究は、英国の科学雑誌『Genome Biology』のオンライン版(1月20日付)に掲載されます。

図 主成分分析(PCA)の性能評価の手順
背景
私たちの臓器には多種多様な細胞が含まれており、これらの細胞が互いに関連して臓器の機能を支えています。しかし、臓器にどのような細胞がどの程度含まれているか、それぞれの細胞がどのように臓器機能を支えているかについては、十分に理解が進んでいません。臓器の疾患の理解や診断、創薬を精緻に行うためには、臓器に含まれる細胞種類[5]やその数、細胞状態[5]やその機能、薬剤に対する応答を調べる必要があります。
細胞が持つ多様な機能は、ゲノムDNAにコードされた数万種類のRNAのうち、どれがどのくらい発現するのかの組み合わせによって決まります。遺伝子は、RNAに転写されたのちさまざまなタンパク質に翻訳され、細胞のさまざまな機能を担います。そのため、臓器を構成する細胞種を判別し、その機能を類推するには、1細胞が持つRNAの量と種類を知る必要があります。これを実現する技術が1細胞RNAシーケンス法(1細胞RNA-seq)です。
1細胞RNA-Seqは、臓器から分離された1細胞をランダムに計測するため、どのデータが何の細胞なのか事前には分かりません。そのため、データ取得後に各種データ解析によって、細胞の種類や数、機能が特定されます。1細胞RNA-Seqのデータは、「遺伝子数×細胞数の次元」を持つ行列で表されます。例えば、ヒト細胞には数万個の遺伝子が存在し、1細胞RNA-Seqでは数千から数百万個の細胞を観測するため、最大で数千億次元のデータとなります。このような大きな次元を持つデータを人間が直感的に理解することは困難です。
そこで、データを低次元に射影して細胞の分布を確認する「次元圧縮法」が大きな役割を果たします。次元圧縮の手法はいくつも存在しますが、その中でも「主成分分析(PCA)」は、あらゆる解析手法の基盤となっています。PCAにより、少ない変数に次元を圧縮することで、データを理解しやすくすることができます。例えば、1細胞RNA-Seqのデータを2次元に圧縮すると、多くの場合、データを細胞が分化してからの時間と細胞種類の二つの次元で説明できます。
しかし、計測細胞数が増加するにつれて、従来のPCAアルゴリズムでは、現実的な計算時間、メモリ使用量で実行できず、データ解析のボトルネックになっていました。また、いくつか高速・軽量なPCAアルゴリズムは提案されているものの、大規模1細胞RNA-Seqデータにおける有効性はよく知られていませんでした。
研究手法と成果
PCAは、遺伝子ごとに平均値を引いたデータ行列の特異値分解(SVD)[6]や、共分散行列[7]の固有値分解(EVD)[8]として定式化されます。多くのPCAのツールでは、データ行列が全てメモリ上に展開されている前提で、SVDやEVDを計算します。しかし、データ数が100万細胞レベルにもなる大規模RNA-Seqでは、行列の全要素を全てメモリ上に載せることは難しくなります。また、多くの研究では、PCAの計算を省略せずに、つまり計算可能な特異値[9]・特異ベクトル[9]を全て計算する(full-rank SVD)ため時間がかかります。
そのため本研究では、データ行列の一部分だけを逐次的に読み込んで計算に利用する実装(Out-of-core)や、特異値が大きい数個の特異値・特異ベクトルのペアのみを計算するアルゴリズム(truncated SVD)を利用することで、巨大な1細胞RNA-seqデータを高速かつ軽量にPCAができるアルゴリズムやソフトウェア実装法、データ形式を報告しました。
共同研究チームはまず、10種類の代表的なPCAアルゴリズムに注目しました。それらを実装したソフトウェアを選定し、ソフトウェアがないアルゴリズムは、Julia言語[10]で独自に実装しました。その結果、21種類の代表的なソフトウェアが選定されました(図1)。次に、これらのソフトウェアの性能を四つの実データと18の人工データ(図1)を用いて比較しました。その結果、一部のデータだけでEVDを行うアルゴリズム(ダウンサンプリング法[11]や確率的勾配法[12]に分類されるアルゴリズム)では、細胞分類の精度を低下させることが分かりました(図1)。
また、計算時間やメモリ使用量はアルゴリズムの違いだけでなく、データ形式やデータの読み込み方にも大きく依存することから、遺伝子発現量がゼロの部分を除いた形式(疎行列フォーマット)を利用すると、計算時間やメモリ使用量の削減効果が高いことが分かりました(図1)。さらに、1細胞RNA-seqのデータは、その検出感度のため遺伝子発現量が計測されない遺伝子が存在します。このような1細胞RNA-seqデータの特徴を踏まえたデータ形式が重要であることが分かりました。
そして、上記の性能評価で利用された一部のPCAアルゴリズムに加え、データの行・列ごとの統計量の計算、それら統計量をもとにした特徴的な発現変動を示す遺伝子や特徴的な細胞を選別する統計量(Highly Variable Genes[13]など)も全て、「Out-of-core」で実行できる実用的なソフトウェアを開発しました(図1)。共同研究チームはJuliaパッケージOnlinePCA.jlをオープンソースソフトウェアとして公開し、誰でも自由に使用できるようにしています。
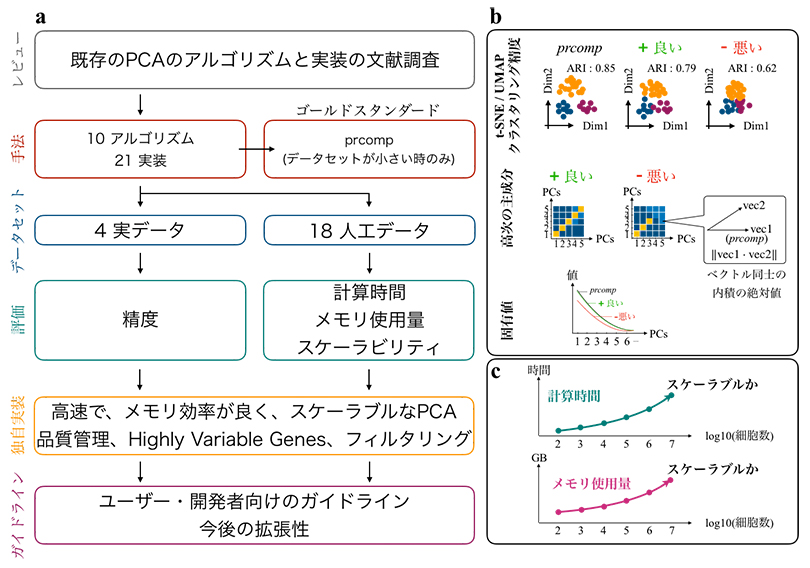
図1 主成分分析(PCA)の性能評価
a)性能評価の手順。大規模なPCAを実行するアルゴリズムを網羅的に調査し実装を収集した。実装されていないものは共同研究チームにより実装された。人工データと実際の1細胞RNA-seqデータを用いて、次元圧縮の性能をさまざまな項目で評価した。ユーザや開発者に向けて次元圧縮法の実装方法についてガイドラインを示した。
b)次元圧縮の性能評価項目。
c)実行速度やメモリ消費量などのスケーラブルさの評価。
今後の期待
本研究では、100万細胞レベルの大規模1細胞RNA-Seqに利用可能な、正確で、高速かつ低メモリ使用量のPCAの実装を選定しました。データ解析で利用されるプログラミング言語は多岐にわたることから、R言語、Python言語、Julia言語において、データ行列のサイズごとに推奨されるPCAの実装のガイドラインを策定しました。
今回のベンチマークで特筆すべき点としては、ランダム特異値分解[14]による近似的なPCAの精度が、実用に耐えうる程度に高かったことです。この手法は1細胞RNA-Seqの分野では利用実績が少ないものの、シンプルで実装しやすいアルゴリズムであることから、今後の利用が期待できます。
また、本研究でのアルゴリズムや実装の工夫は、1細胞RNA-Seqデータで利用される他のさまざまなアルゴリズムでも参考になります。例えば、遺伝子や細胞のクラスタリングや、複数のデータ行列の同時分解によるバッチエフェクト除去[15]、多次元配列(テンソル)の分解[16]による細胞間相互作用検出などで利用されるアルゴリズムの高速化・軽量化への貢献が期待できます。
補足説明
1.主成分分析(PCA)
多変量データの次元を削減し、特徴的に変動している因子を発見する統計手法。相関のある多数の変数からなるデータを、互いに相関のない少数の変数によって説明する。PCAはPrincipal Component Analysisの略。
2.1細胞RNAシーケンス法(1細胞RNA-seq)
1細胞中に含まれるRNAをDNAシーケンサーで配列決定し、網羅的かつ定量的にその量や種類を決定する方法。微量なRNAを用いるため、微量RNAから相補的DNA(cDNA)を合成する「逆転写反応」と、シーケンス可能な量までcDNAを増幅させる「全cDNA増幅法」の二つのステップからなる。
3.疎行列フォーマット
行列に含まれる要素のうち、非ゼロの値とその値の行列上の位置のみを保存したファイル形式。十分に行列が疎(ゼロが多い)の場合に、計算速度や、メモリ使用量の効率化に寄与する。
4.Out-of-core
データ行列の全要素をメモリに読み込むのではなく、一部分だけを切り出して、逐次的に計算に利用するタイプの実装。オンライン、逐次的、オンディスクといった呼ばれ方もする。
5.細胞種類、細胞状態
多細胞生物は、たくさんの細胞から構成される。それぞれの細胞は役割、すなわち機能を持つ。例えば、免疫を担う細胞や角膜のように透明な細胞、神経活動を支える電気や化学物質で他の細胞と通信する細胞などがある。また、同じ細胞でもその状態が異なる場合がある。免疫の細胞でも機能が未熟なものから、成熟して機能を発揮する細胞まである。
6.特異値分解(SVD)
行列X(X: N×M、N < Mとする)を、X =(N×N行列)×(N×Nの対角行列)×(N×M行列)のように分解するアルゴリズム。SVDはSingular Value Decompositionの略。
7.共分散行列
二つのデータのばらつき具合を示すのが共分散である。行列には複数のデータが含まれるが、これらの二つのデータ間の共分散を行列にまとめたものを共分散行列と呼ぶ。行列に対角成分は自分のデータの分散、それ以外の成分にはそれぞれの共分散が含まれる。
8.固有値分解(EVD)
正方行列X(X: N×N)を、X =(N×N行列)×(N×Nの対角行列)のように分解するアルゴリズム。EVDはEigen Value Decompositionの略。
9.特異値、特異ベクトル
特異値分解により、行列X(X: N×M、N < Mとする)を、X =(N×N行列)×(N×Nの対角行列)×(N×M行列)に分解した際のN×N行列の対角成分の値を特異値、N×N行列とN×M行列をそれぞれ左特異ベクトル、右特異ベクトルと呼ぶ。
10.Julia言語
2012年に公開された比較的新しいプログラミング言語。高速に動作し、科学技術計算に使われるさまざまな機能を標準で備えるため、ライフサイエンスのみならず、データサイエンス分野で広く利用されつつある。
11.ダウンサンプリング法
ランダムに選んだ遺伝子の発現量のみを使って、特異値分解を計算する手法。選ぶ遺伝子の数を少なくすることで、データ行列の全要素をメモリ上に載せることが可能となるが、今回のベンチマークにより、その分精度が下がる可能性があることが明らかになった。
12.確率的勾配法
ある目的関数の最大値(または最小値)を求める最適化問題において、適当な初期値をもとに、目的関数の傾きが急な方向に逐次的に解を更新していく最適化手法の一つ。特に、PCAにおける確率的勾配法はOjaの手法、Generalized Hebbian Algorithmとして知られている。
13.Highly Variable Genes
平均値に対して分散(散らばり)が大きい遺伝子を求める仮説検定手法。p値が小さい遺伝子のみを利用することで、経験的に次元圧縮やクラスタリングの精度が向上することが多いことが知られている。
14.ランダム特異値分解
truncated SVDの一種。高次元データをランダムに低次元に射影してから、各種演算を行うため、高速かつ低メモリに計算ができる。
15.バッチエフェクト除去
実験環境(機器や観測者など)の違いで生じるデータ変動をバッチエフェクトという。1細胞RNA-Seqデータでは、多くの場合、細胞の種類・状態の違いによる変動よりも、バッチエフェクトの方が大きくなることが問題となっている。データに含まれているバッチエフェクトを回避して、生物学的な変動のみに注目するために、SeuratやLIGARといったソフトウェアの内部では、Multi-CCA、joint NMFなど、PCAとよく似た複数の行列同時分解アルゴリズムが実行される。
16.テンソル分解
行列が行(縦)、列(横)に値が配置されているのに対して、奥行きを加え、より高次なデータ構造にしたものをテンソルという。scTensorというソフトウェアでは、細胞間相互作用をテンソルとして表現し、PCAと同様に分解アルゴリズムで、少数のパターンを検出する。
共同研究チーム
理化学研究所 生命機能科学研究センター バイオインフォマティクス研究開発チーム
特別研究員 露崎 弘毅(つゆざき こうき)
リサーチアソシエイト 佐藤 建太(さとう けんた)
チームリーダー 二階堂 愛(にかいどう いとし)
京都大学大学院 情報学研究科
特定准教授 佐藤 寛之(さとう ひろゆき)
研究支援
本研究の一部は、日本学術振興会(JSPS)科学研究費補助金若手研究「非負値テンソル分解を用いたオーファン受容体結合リガンドの同定及び機能解明(研究代表者:露崎 弘毅)」、科学技術振興機構(JST)戦略的創造研究推進事業(CREST)「[1細胞] 統合1細胞解析のための革新的技術基盤(研究統括:菅野純夫)」の研究課題「臓器・組織内未知細胞の命運・機能の1細胞オミクス同時計測(研究代表者: 二階堂愛)」、JSTおよび日本医療研究開発機構(AMED)再生医療実現拠点ネットワークプログラム「超多検体オミクスによる細胞特性の計測(研究代表者: 二階堂愛)」「iPS・分化細胞集団の不均質性を1細胞・全遺伝子解像度で高速に測定する技術の開発(研究代表者: 二階堂愛)」の支援を受けて行われました。
原論文情報
Koki Tsuyuzaki, Hiroyuki Sato, Kenta Sato, Itoshi Nikaido, “Benchmarking principal component analysis for large-scale single-cell RNA-sequencing”, Genome Biology, 10.1186/s13059-019-1900-3
発表者
理化学研究所
生命機能科学研究センター バイオインフォマティクス研究開発チーム
特別研究員 露崎 弘毅(つゆざき こうき)
チームリーダー 二階堂 愛(にかいどう いとし)
報道担当
理化学研究所 広報室 報道担当

