
2024-08-22 カリフォルニア工科大学(Caltech)
")
To render the samarium diiodide active, the chemists added an acid to the reaction. In this picture, varying amounts of acid have been added, with the strongest acid on the left and weakest acid on the right. The stronger acids worked faster, hence the vials to the left have more of the active (purple) form of the samarium reagent.Credit: Caltech/Chungkeun Shin
<関連情報>
- https://www.caltech.edu/about/news/teaching-an-old-metal-new-tricks
- https://www.science.org/doi/10.1126/science.adp5777
- https://www.nature.com/articles/s41586-022-05011-6
SmIII-アルコキシドプロトノリシスが可能にする還元的サマリウム(電気)触媒作用 Reductive samarium (electro)catalysis enabled by SmIII-alkoxide protonolysis
Emily A. Boyd, Chungkeun Shin, David J. Charboneau, Jonas C. Peters, and Sarah E. Reisman
Science Published:22 Aug 2024
DOI:https://doi.org/10.1126/science.adp5777
Editor’s summary
Samarium diiodide is widely used as a reductant in reactions of carbonyl compounds. However, it has poor solubility and must generally be used in excess quantities. Attempts to use this reductant catalytically have been hindered by its tight binding to oxygen during reactions. Boyd et al. now report that a well-chosen acid can help cleave the samarium-oxygen bond and induce turnover with electrons provided electrochemically or by metallic zinc. Lactone formation from ketone coupling with acrylates showcased the catalysis. —Jake S. Yeston
Abstract
Samarium diiodide (SmI2) is a privileged, single-electron reductant deployed in diverse synthetic settings. However, generalizable methods for catalytic turnover remain elusive because of the well-known challenge associated with cleaving strong SmIII–O bonds. Prior efforts have focused on the use of highly reactive oxophiles to enable catalyst turnover. However, such approaches give rise to complex catalyst speciation and intrinsically limit the synthetic scope. Herein, we leveraged a mild and selective protonolysis strategy to achieve samarium-catalyzed, intermolecular reductive cross-coupling of ketones and acrylates with broad scope. The modularity of our approach allows rational control of selectivity based on solvent, pKa (where Ka is the acid dissociation constant), and the samarium coordination sphere and provides a basis for future developments in catalytic and electrocatalytic lanthanide chemistry.
プロトン結合電子移動によるタンデム電極触媒的N2固定化反応 Tandem electrocatalytic N2 fixation via proton-coupled electron transfer
Pablo Garrido-Barros,Joseph Derosa,Matthew J. Chalkley & Jonas C. Peters
Nature Published:31 August 2022
DOI:https://doi.org/10.1038/s41586-022-05011-6
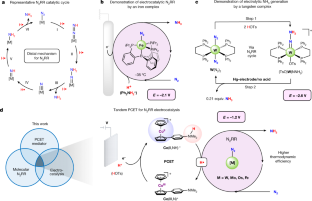
Abstract
New electrochemical ammonia (NH3) synthesis technologies are of interest as a complementary route to the Haber–Bosch process for distributed fertilizer generation, and towards exploiting ammonia as a zero-carbon fuel produced via renewably sourced electricity1. Apropos of these goals is a surge of fundamental research targeting heterogeneous materials as electrocatalysts for the nitrogen reduction reaction (N2RR)2. These systems generally suffer from poor stability and NH3 selectivity; the hydrogen evolution reaction (HER) outcompetes N2RR3. Molecular catalyst systems can be exquisitely tuned and offer an alternative strategy4, but progress has been thwarted by the same selectivity issue; HER dominates. Here we describe a tandem catalysis strategy that offers a solution to this puzzle. A molecular complex that can mediate an N2 reduction cycle is partnered with a co-catalyst that interfaces the electrode and an acid to mediate proton-coupled electron transfer steps, facilitating N−H bond formation at a favourable applied potential (−1.2 V versus Fc+/0) and overall thermodynamic efficiency. Certain intermediates of the N2RR cycle would be otherwise unreactive via uncoupled electron transfer or proton transfer steps. Structurally diverse complexes of several metals (W, Mo, Os, Fe) also mediate N2RR electrocatalysis at the same potential in the presence of the mediator, pointing to the generality of this tandem approach.
")
")