
- MeV-UEDで未踏の領域に挑む 分子イオンの誕生と構造進化を解明
Delving into Unexplored Realms, MeV-UED Illuminates the Birth and Structural Evolution of a Molecular Ion
- 溶液中の過渡的分子構造の超高速X線回折 Ultrafast X-ray Diffraction of Transient Molecular Structures in Solution
- 結合形成を媒介する分子振動の出現をマッピングする Mapping the emergence of molecular vibrations mediating bond formation
- フェムト秒X線散乱による溶液中の結合形成の直接観察 Direct observation of bond formation in solution with femtosecond X-ray scattering
- 溶液中の過渡的分子構造の超高速X線回折 Ultrafast X-ray Diffraction of Transient Molecular Structures in Solution
MeV-UEDで未踏の領域に挑む 分子イオンの誕生と構造進化を解明 Delving into Unexplored Realms, MeV-UED Illuminates the Birth and Structural Evolution of a Molecular Ion
2024-01-11 韓国基礎科学研究院(IBS)
◆研究は1,3-ジブロモプロパンのカチオンに焦点を当て、安定な「ダークステート」の後に環状構造の中間体に変化する現象を明らかにしました。この成功は信号処理、モデリング分析、REMPI技術の適用によるもので、安定なリング状分子の生成を解明しました。この突破口は、イオン化学、化学反応、物質特性、宇宙化学などに影響を与え、選択的なイオンの構造ダイナミクスの初のリアルタイム観察を成し遂げました。
<関連情報>
- https://www.ibs.re.kr/cop/bbs/BBSMSTR_000000000738/selectBoardArticle.do
- https://www.nature.com/articles/s41586-023-06909-5
- https://www.nature.com/articles/s41586-020-2417-3
- https://www.nature.com/articles/nature14163
- https://www.science.org/doi/10.1126/science.1114782
溶液中の過渡的分子構造の超高速X線回折 Ultrafast X-ray Diffraction of Transient Molecular Structures in Solution
Jun Heo,Doyeong Kim,Alekos Segalina,Hosung Ki,Doo-Sik Ahn,Seonggon Lee,Jungmin Kim,Yongjun Cha,Kyung Won Lee,Jie Yang,J. Pedro F. Nunes,Xijie Wang & Hyotcherl Ihee
Nature Published:10 January 2024
DOI:https://doi.org/10.1038/s41586-023-06909-5
")
Abstract
Molecular ions are ubiquitous and play pivotal roles1,2,3 in many reactions, particularly in the context of atmospheric and interstellar chemistry4,5,6. However, their structures and conformational transitions7,8, particularly in the gas phase, are less explored than those of neutral molecules owing to experimental difficulties. A case in point is the halonium ions9,10,11, whose highly reactive nature and ring strain make them short-lived intermediates that are readily attacked even by weak nucleophiles and thus challenging to isolate or capture before they undergo further reaction. Here we show that mega-electronvolt ultrafast electron diffraction (MeV-UED)12,13,14, used in conjunction with resonance-enhanced multiphoton ionization, can monitor the formation of 1,3-dibromopropane (DBP) cations and their subsequent structural dynamics forming a halonium ion. We find that the DBP+ cation remains for a substantial duration of 3.6 ps in aptly named ‘dark states’ that are structurally indistinguishable from the DBP electronic ground state. The structural data, supported by surface-hopping simulations15 and ab initio calculations16, reveal that the cation subsequently decays to iso-DBP+, an unusual intermediate with a four-membered ring containing a loosely bound17,18 bromine atom, and eventually loses the bromine atom and forms a bromonium ion with a three-membered-ring structure19. We anticipate that the approach used here can also be applied to examine the structural dynamics of other molecular ions and thereby deepen our understanding of ion chemistry.
結合形成を媒介する分子振動の出現をマッピングする Mapping the emergence of molecular vibrations mediating bond formation
Jong Goo Kim,Shunsuke Nozawa,Hanui Kim,Eun Hyuk Choi,Tokushi Sato,Tae Wu Kim,Kyung Hwan Kim,Hosung Ki,Jungmin Kim,Minseo Choi,Yunbeom Lee,Jun Heo,Key Young Oang,Kouhei Ichiyanagi,Ryo Fukaya,Jae Hyuk Lee,Jaeku Park,Intae Eom,Sae Hwan Chun,Sunam Kim,Minseok Kim,Tetsuo Katayama,Tadashi Togashi,Sigeki Owada,Makina Yabashi,Sang Jin Lee,Seonggon Lee,Chi Woo Ahn,Doo-Sik Ahn,Jiwon Moon,Seungjoo Choi,Joonghan Kim,Taiha Joo,Jeongho Kim,Shin-ichi Adachi &Hyotcherl Ihee
Nature Published:24 June 2020
DOI:https://doi.org/10.1038/s41586-020-2417-3
Abstract
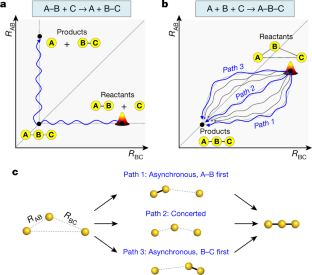
Fundamental studies of chemical reactions often consider the molecular dynamics along a reaction coordinate using a calculated or suggested potential energy surface1,2,3,4,5. But fully mapping such dynamics experimentally, by following all nuclear motions in a time-resolved manner—that is, the motions of wavepackets—is challenging and has not yet been realized even for the simple stereotypical bimolecular reaction6,7,8: A–B + C → A + B–C. Here we track the trajectories of these vibrational wavepackets during photoinduced bond formation of the gold trimer complex [Au(CN)2−]3 in an aqueous monomer solution, using femtosecond X-ray liquidography9,10,11,12 with X-ray free-electron lasers13,14. In the complex, which forms when three monomers A, B and C cluster together through non-covalent interactions15,16, the distance between A and B is shorter than that between B and C. Tracking the wavepacket in three-dimensional nuclear coordinates reveals that within the first 60 femtoseconds after photoexcitation, a covalent bond forms between A and B to give A–B + C. The second covalent bond, between B and C, subsequently forms within 360 femtoseconds to give a linear and covalently bonded trimer complex A–B–C. The trimer exhibits harmonic vibrations that we map and unambiguously assign to specific normal modes using only the experimental data. In principle, more intense X-rays could visualize the motion not only of highly scattering atoms such as gold but also of lighter atoms such as carbon and nitrogen, which will open the door to the direct tracking of the atomic motions involved in many chemical reactions.
フェムト秒X線散乱による溶液中の結合形成の直接観察 Direct observation of bond formation in solution with femtosecond X-ray scattering
Kyung Hwan Kim,Jong Goo Kim,Shunsuke Nozawa,Tokushi Sato,Key Young Oang,Tae Wu Kim,Hosung Ki,Junbeom Jo,Sungjun Park,Changyong Song,Takahiro Sato,Kanade Ogawa,Tadashi Togashi,Kensuke Tono,Makina Yabashi,Tetsuya Ishikawa,Joonghan Kim,Ryong Ryoo,Jeongho Kim,Hyotcherl Ihee &Shin-ichi Adachi
Nature Published:18 February 2015
DOI:https://doi.org/10.1038/nature14163
Abstract
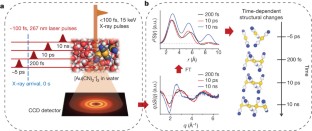
The making and breaking of atomic bonds are essential processes in chemical reactions. Although the ultrafast dynamics of bond breaking have been studied intensively using time-resolved techniques1,2,3, it is very difficult to study the structural dynamics of bond making, mainly because of its bimolecular nature. It is especially difficult to initiate and follow diffusion-limited bond formation in solution with ultrahigh time resolution. Here we use femtosecond time-resolved X-ray solution scattering to visualize the formation of a gold trimer complex[Au(CN)–2]3 in real time without the limitation imposed by slow diffusion. This photoexcited gold trimer, which has weakly bound gold atoms in the ground state4,5,6, undergoes a sequence of structural changes, and our experiments probe the dynamics of individual reaction steps, including covalent bond formation, the bent-to-linear transition, bond contraction and tetramer formation with a time resolution of ∼500 femtoseconds. We also determined the three-dimensional structures of reaction intermediates with sub-ångström spatial resolution. This work demonstrates that it is possible to track in detail and in real time the structural changes that occur during a chemical reaction in solution using X-ray free-electron lasers7 and advanced analysis of time-resolved solution scattering data.
溶液中の過渡的分子構造の超高速X線回折 Ultrafast X-ray Diffraction of Transient Molecular Structures in Solution
H. Ihee ,M. Lorenc,T. K. Kim,Q. Y. Kong,M. Cammarata,J. H. Lee,S. Bratos,and M. Wulff
Science Published:19 Aug 2005
DOI:https://doi.org/10.1126/science.1114782
Abstract
We report direct structural evidence of the bridged radical (CH2ICH2·) in a polar solution, obtained using time-resolved liquid-phase x-ray diffraction. This transient intermediate has long been hypothesized to explain stereo-chemical control in many association and/or dissociation reactions involving haloalkanes. Ultrashort optical pulses were used to dissociate an iodine atom from the haloethane molecule (C2H4I2) dissolved in methanol, and the diffraction of picosecond x-ray pulses from a synchrotron supports the following structural dynamics, with ∼0.01 angstrom spatial resolution and ∼100 picosecond time resolution: The loss of one iodine atom from C2H4I2 leads to the C-I-C triangular geometry of CH2ICH2·. This transient C2H4I then binds to an iodine atom to form a new species, the C2H4I-I isomer, which eventually decays into C2H4 + I2. Solvent dynamics were also extracted from the data, revealing a change in the solvent cage geometry, heating, and thermal expansion.


")