2021-07-19 東京大学
○発表者:
溝口 照康(東京大学 生産技術研究所 教授)
鈴木 叡輝(研究当時:東京大学 大学院工学系研究科 修士課程2年)
柴田 基洋(東京大学 生産技術研究所 助教)
○発表のポイント:
◆化学反応や吸着といった現象は、原子が他の原子や分子などと結合する過程が組み合わさって生じます。結合強度や距離などの結合物性を知るには、それぞれの結合についてモデルを作成してシミュレーションする必要があります。
◆今回、原子―原子、原子―エチレン分子、原子―グラフェンという比較的単純な化学結合を対象とし、結合「前」の状態で得られる情報だけで、結合「後」の結合物性を高精度に予測できる人工知能を構築しました。また、高精度の予測には、結合を形成する原子、分子、固体の個々の状態(状態密度)の情報が重要であることを明らかにしました。さらに、開発した手法を用いることで、わずかなデータ量の学習で十分な精度を実現できることも明らかになりました。
◆今後、分子―分子や、分子―固体など、より複雑な化学結合の予測へと応用されることで、吸着や化学反応の予測に役立ち、物質開発のさらなる加速が期待されます。
○発表概要:
東京大学 生産技術研究所の溝口 照康 教授、東京大学 大学院工学系研究科 修士課程2年の鈴木 叡輝 大学院生(研究当時)、東京大学 生産技術研究所の柴田 基洋 助教らの研究グループは、化学結合(注1)を高精度に予測することができる記述子(注2)を発見しました。
原子が他の原子や、分子、固体表面に近づくと、それぞれが持っている軌道が相互作用し、時としてお互いの電子を共有・移動し「化学結合」が形成されます。お互いの原子の大きさや、共有・移動する電子の量によって、化学結合の強度や距離が決まります。化学結合の形成は、吸着や化学反応のきっかけとなるため、その理解は物質開発において重要です。
一方で、化学結合の強度や距離などの結合物性は、結合をつくる原子―原子、原子―分子、原子―固体などの組み合わせによって異なります。結合物性を知るためには、結合のモデルを作成し、計算機を使って数分から数時間かけてシミュレーション(注3)する必要があります。さらに、さまざまな種類の結合物性を知るには、それらすべてに対して同様なシミュレーションを行う必要があります。
そこで、そのようなシミュレーションを行わずに、結合する「前」から得られる情報だけで、結合物性を予測するための人工知能の構築を目指しました。本研究では入力層と出力層が脳を模した多層のネットワークでつながれている「ニューラルネットワーク(注4)」を利用しました。結合する「前」の情報だけで、化学結合を予測する人工知能を構築することを目指し、入力データに、結合する「前」、つまり原子や分子が孤立している状態の情報を、出力データに、結合した「後」の結合物性を利用してネットワークを築きました。
本研究では、結合を形成する原子、分子、固体の個々の状態、つまり結合する「前」の状態における状態密度(注5)を入力データに用いることで、結合物性を高精度に予測できることを明らかにしました。このことは、結合する「前」の状態密度が、結合物性を予測するための優れた記述子であることを示しています。また、原子―原子に加えて、原子―分子、原子―固体の結合強度、結合距離、フェルミ準位、共有結合電荷を高精度に予測するモデルを構築することに成功しました。
同成果の概念図を図1に示します。本研究では、人工知能技術により、様々な化学結合を高速かつ高精度に予測することに成功しました。化学結合の形成は、吸着や化学反応などのきっかけとして重要です。そのような化学結合を高精度に予測できる記述子を発見した本研究により、物質開発が加速できると期待されます。
本研究成果は令和3年7月19日に日本応用物理学会の速報国際誌の「Applied Physics Express」オンライン版に掲載されました。
○発表内容:
<研究背景>
原子が他の原子や、分子、固体表面に近づくことで、それぞれの軌道が相互作用し、時としてお互いの電子を移動・共有することで「化学結合」が形成されます。お互いの原子の大きさや、共有する電子の量によって、化学結合の強さや、結合距離が決まります。そのような化学結合の形成は、吸着反応や化学反応のきっかけとなるため、それを理解することは物質開発において重要です。
一方で、化学結合の強さや、結合距離、共有される電子の数などの結合物性は、化学結合を作る組み合わせによって異なります。そのような結合物性を知るためには、目的の結合の組み合わせの原子や分子、固体表面を含むモデルを作成し、第一原理計算(注6)によってシミュレーションする必要があります。
計算する前では、結合距離がいくつになるかは分からないため、原子半径などから適当な位置に原子を置き、計算の過程で結合距離を最適化していきます。そのような最適化には、数分から数時間を要します。さらに、さまざまな分子や表面の結合物性を知るためには、目的の結合の組み合わせすべてに対して、同様なシミュレーションを実施する必要がありました。
<研究内容>
本研究で検討した化学結合の種類を図2に示します。まず、周期表内のH(水素)からRn(ラドン)までの原子について、それら原子がつくる2原子分子における化学結合の形成挙動と、それらの原子とエチレン分子、さらにグラフェンとの間の化学結合の形成挙動を網羅的に計算しました。
結合物性を予測するために人工知能で使用されているニューラルネットワークを利用しました。ニューラルネットワークでは、入力層と出力層が脳を模した多層のネットワークでつながれています。本研究では、図3に模式的に示すように、入力データに結合する「前」、つまり孤立している状態の情報を、出力データには結合した「後」の、結合強度や結合距離などの結合物性を利用してネットワークを築きました。そうすることで、結合する「前」の情報だけで、化学結合を予測する人工知能を構築することができます。
その結果、結合する「前」の状態密度を記述子として使用することで、結合強度を0.22eV(エレクトロンボルト)、結合距離を0.011ナノメートルの誤差で予測することができました。これは結合する「前」の状態密度が、化学結合を予測するための優れた記述子であることを示しています。
さらに、本研究では、化学結合を予測するために必要な学習データの量も調べました。一般的な機械学習の研究では、予測するデータの量に対して、約3~4倍程度のデータ量が学習に使用されています。しかし、本研究では、予測データ量のわずか1/5程度という少ないデータ量で学習しても、十分な精度で化学結合を予測できることも明らかにしました。本研究では、人工知能技術により、様々な化学結合を高速かつ高精度に予測することに成功しました。同成果の概念図を図1に示します。
<今後の展開>
化学結合の形成は、吸着や化学反応などの素過程として重要です。本研究では、原子―原子、原子―エチレン分子、原子―グラフェンという比較的単純な化学結合を対象としました。今後、分子―分子や、分子―固体等のより複雑な化学結合を予測することができれば、吸着や化学反応の予測にも役立ち、物質開発のさらなる加速が期待されます。
○発表雑誌:
雑誌名:「Applied Physics Express」(オンライン版:日本時間7月19日午後6時 掲載予定)
論文タイトル:Accurate Prediction of Bonding Properties by A Machine Learning-based Model using Isolated States Before Bonding(結合前の情報により作製した機械学習モデルによる結合物性の高精度予測)
著者:Eiki Suzuki, Kiyou Shibata, and Teruyasu Mizoguchi(鈴木叡輝、柴田基洋、溝口 照康)
DOI: 10.35848/1882-0786/ac083b
○問い合わせ先:
東京大学 生産技術研究所
教授 溝口 照康(みぞぐち てるやす)
○用語解説:
注1)化学結合
原子間で形成される結合。各原子が持つ軌道の相互作用により電子を共有したり移動したりして形成される。化学結合の強さや結合距離は、各原子のサイズや共有・移動する電子の量によって異なる。
注2)記述子
機械学習のモデルを構築するための入力データ。特徴量や説明変数ともよばれている。本研究では、注4)のニューラルネットワークの入力データ。
注3)シミュレーション
本研究のシミュレーションは、量子化学理論に基づく第一原理計算のこと。電子の状態を計算することで、化学結合の強さ、結合距離、電子の移動・共有量などを計算することができる。また、計算する過程において、最適な原子配置や結合距離を計算してくれる。計算時間は、計算する原子の数にも依存する。二原子分子であれば数分で終了するが、原子と固体の化学結合を計算するためには、構造の最適化に時間を要し、数時間かかることもある。
注4)ニューラルネットワーク
脳を模した機械学習の手法で、入力データと出力データの間を多層のネットワークでつなぐ方法。本研究では、入力データが基底状態のスペクトルで、出力データが励起状態のスペクトルとなっている。教師あり学習によりネットワークのつなぎ方を変え、出力データの予測精度をあげることができる。
注5)状態密度
注3)のシミュレーションによって得られる計算結果の一種。原子や分子などが有する軌道のエネルギーや、軌道の数に関する情報を有している。結合をつくる「前」と「後」とでは当然異なる状態密度が得られるが、本研究では、結合をつくる「前」の状態密度が有効な記述子であることを明らかにした。
注6)第一原理計算
量子力学に基づき、原子や電子の状態を計算する手法。化学結合の計算に加えて、構造の最適化も可能。
○添付資料:

図1 本研究の概念図。今回構築した人工知能(中央)を用いることで、さまざまな種類の結合(Chemical bonding)を予測(prediction)することに成功しました。
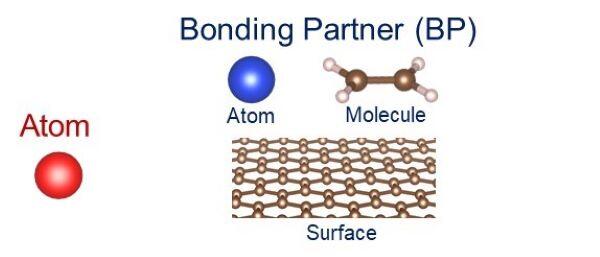
図2 原子(左側)と、右側の結合相手(Bonding partner:BP)。BPには、異なる原子と、エチレン分子、グラフェン表面を用いた。
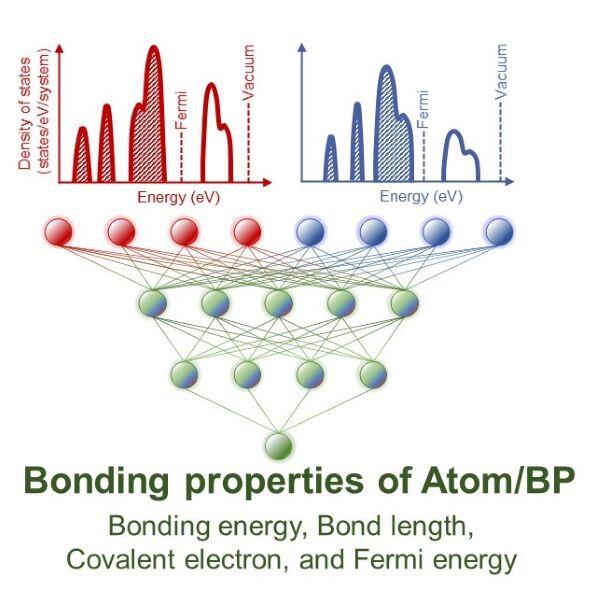
図3 本研究で用いたニューラルネットワークの入力値と出力の模式図。入力値には、図2で示した原子(図2左側の赤球)と、BP(図2右側の原子、分子、表面)の状態密度(Density of states)を用いた。出力は結合強度、結合距離、共有電子数、およびフェルミ準位などの結合物性。

