
2024-10-26 東京大学
発表のポイント
- 第一原理計算の結果を教師データとして液体分子系の双極子モーメントを予測する新しいニューラルネットワーク手法を開発し、高速かつ高精度な誘電特性の計算を可能にした。
- 多くの分子系に適用できる汎用的な枠組みであり、液体メタノール・エタノールへの適用によってその精度の高さを実証した。
- 次世代高速移動通信6Gに必要な低誘電材料などの誘電特性計算に応用することで、その材料開発を加速する効果が期待される。

発表概要
東京大学大学院理学系研究科の天野智仁大学院生(当時)、常行真司教授は、JSR株式会社と東京大学の包括連携拠点CURIEにおける共同研究によって、液体分子系に汎用的に適用可能な双極子モーメントの機械学習スキームを開発しました。分子系では化学結合付近に電子が存在するという性質を利用し、各化学結合に対して第一原理計算(注1)によって計算された電子位置を表すワニエセンター(注2)光をニューラルネットワーク(注3)で予測させることで、高精度に双極子モーメントを予測できることを示しました。先行研究では、分子単位で双極子モーメントを予測させるのが主流でしたが、化学結合単位でニューラルネットワークモデルを作成することで、大きな分子系でも精度を落とさずに予測できます。本研究では、開発した手法をアルコールとして重要な液体メタノールおよびエタノールに適用し、誘電スペクトルを高精度で計算可能であることを実証しました。今回開発した手法は、今後さらにポリマーなどの誘電材料開発の加速につながると期待されます。
発表内容
通常は電気を流さないものの、電場を加えると電気を溜める性質を持つ誘電体は、コンデンサーや回路基板など、日常生活の多くの場面で使われています。誘電体の特性や性能は誘電関数という物理量によって決まるため、物質の誘電関数を精度良くシミュレーションすることで材料設計に繋げることができます。誘電関数は電場の周波数ごとに物質がどれくらい双極子モーメントを発生させるかを表しており、その詳細は物質内の原子や電子の運動によって決まります。特にテラヘルツから赤外と呼ばれる周波数領域では主に原子の振動が重要ですが、原子が電子雲を纏って運動するため電子の効果も取り入れることが精確なシミュレーションのために重要です。
現在誘電関数に用いられている分子動力学法(注4)では、あらかじめイオンの電荷を固定した経験的力場による方法が幅広く用いられています。この方法は高速な計算が可能ですが、電荷が固定されているが故に物質によっては双極子モーメントを定量的に再現することが難しい場合があります。一方、原子にかかる力を量子力学に基づいた第一原理計算で計算する第一分子動力学法も主要な方法です。この手法では、双極子モーメントを量子力学的に定式化した現代分極理論(注5)を利用することで、物質の双極子モーメントも高精度で計算できるのが大きなメリットです。双極子モーメントの計算にはワニエセンターの情報があれば良いことが理論的に知られており、第一原理分子動力学法ではこれら二つの理論を用いて双極子モーメントの時間発展を調べることができます。しかしながら計算コストが高く、大規模な系や長時間の計算は難しいという課題があります。
そこで本研究では、第一原理計算によって計算した多数の構造とワニエセンターの情報を教師データとしてニューラルネットワークモデルを構成し、第一原理計算よりも大幅に高速な双極子モーメントの計算を可能にしました。分子系では、ワニエセンターは原子間の化学結合に割り当てることができます。この性質を利用し、化学結合ごとにワニエセンターを予測する機械学習モデルを作成しました(図1)。従来は分子ごとに双極子モーメントを予測する方法が多く用いられていましたが、我々の開発した手法は予測単位をより細かく分割することで分子が大きい場合にも精度が落ちにくいという特徴があります。
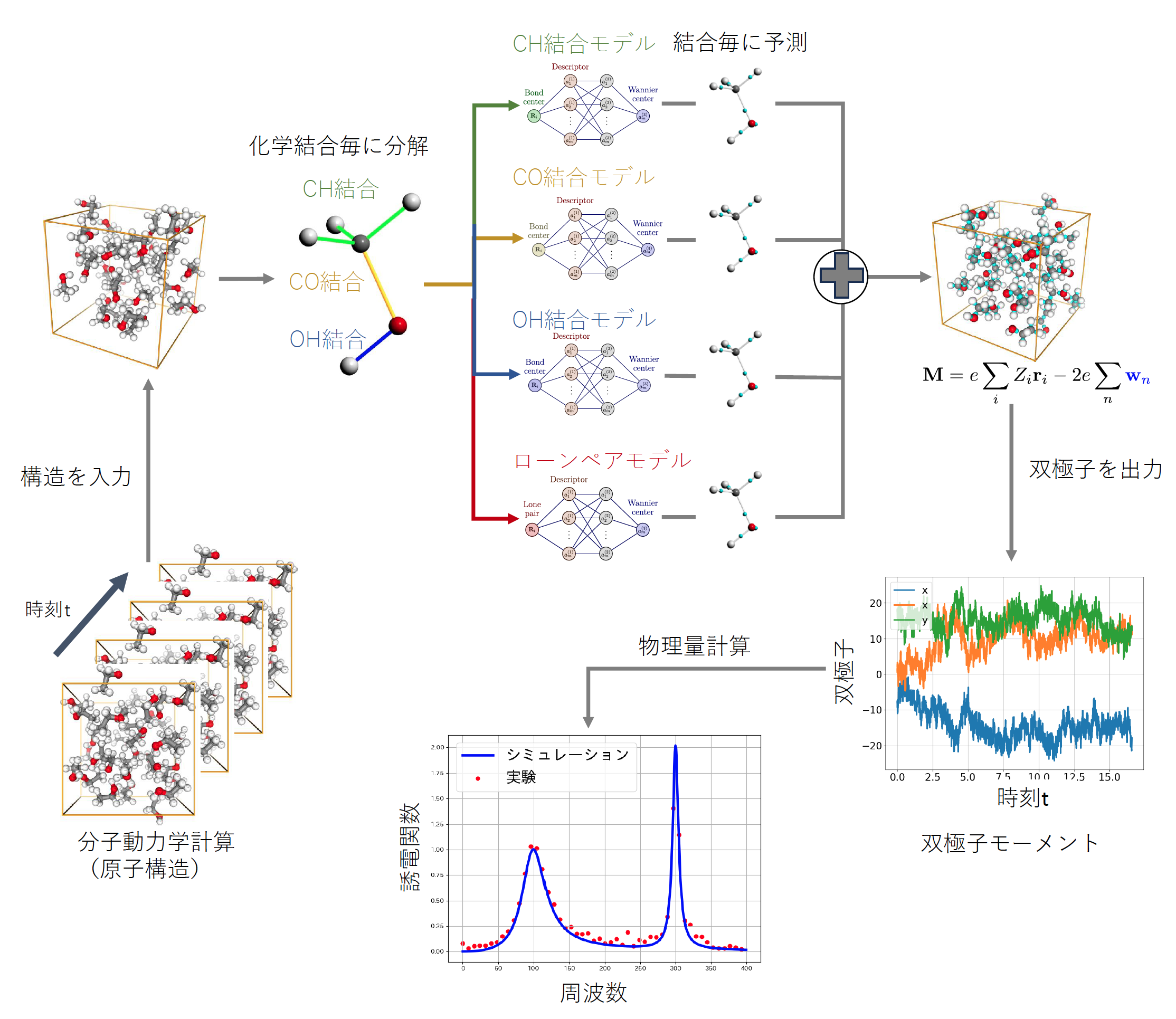
図1.手法の概要:
液体構造から各分子を取り出し、結合の種類ごとに別のニューラルネットワークモデルを利用してワニエセンターの座標を予測する。系全体の双極子モーメントは、原子位置と個別に予測されたワニエセンターから計算される。
我々は開発した手法を液体メタノールおよび液体エタノールに適用しました。その結果、第一原理計算によって計算された液体構造の双極子モーメントを非常によく再現できることがわかりました。さらに、テラヘルツ帯の吸収スペクトルを分析し、液体中では分子間の相互作用によるワニエセンターの移動によって、スペクトルが大きく変化する様子を捉えることに成功しました(図2)。
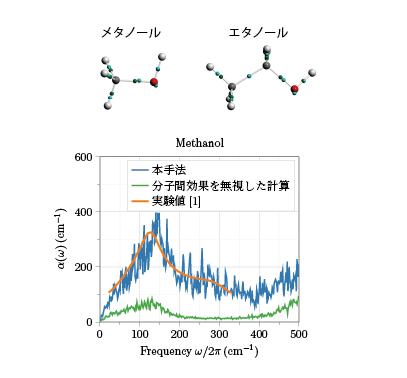
図2.液体メタノールの吸収スペクトル
上)メタノール分子とエタノール分子のワニエセンターのイメージ。赤、灰、白の丸が酸素、炭素、水素原子を表し、各化学結合の中点(緑)からずれた位置にワニエセンター(水色)が現れる。
下)開発した手法で計算した液体メタノールの吸収スペクトル(赤)と、分子間の相互作用を無視して計算した吸収スペクトル(青)。分子間の効果を精確に取り込むことで、スペクトルが大きく増大し、実験と定量的に一致するようになる。実験値[1]はS. Sarkar, D. Saha, S. Banerjee, A. Mukherjee, and P. Mandal, Chemical Physics Letters 678, 65 (2017)より引用。
今回の研究で開発した双極子モーメントのニューラルネットワークモデルは、液体分子系の双極子モーメントを精度よく計算でき、誘電特性の高精度シミュレーションに利用できることが示されました。また、その計算コストの軽さから、従来第一原理計算が適用できなかった大きさの系や、長時間の計算が可能になりました。今後はポリマーなどの高分子系への適用を推し進め、さらに、大規模データを用いて多くの分子系に汎用的に活用可能な機械学習モデルを構築することで、低誘電ポリマー材料などの誘電体材料開発に直接活用でき、革新的な誘電材料開発につながることが期待されます。
東京大学大学院理学系研究科物理学専攻とJSR株式会社は、物理と化学の融合を目指す包括的連携に合意し、2020年4月1日より共同研究を開始いたしました。本研究は、その協創オフィス「JSR・東京大学協創拠点CURIE」における共同研究で得られた成果となっています。[1] S.Sarkar, D. Saha, S. Banerjee, A. Mukherjee, and P. Mandal, Chemical Physics Letters 678, 65 (2017)
論文情報
- 雑誌名
Physical Review B論文タイトル
Chemical bond based machine learning model for dipole moment: Application to dielectric properties of liquid methanol and ethanol著者
Tomohito Amano*, Tamio Yamazaki, Shinji Tsuneyuki(*責任著者)
研究助成
本研究は、JST未来社会創造事業 JPMJMI20A1、 文部科学省 光・量子飛躍フラッグシッププログラム(Q-LEAP)JPMXS0118067246の支援により実施されました。また、本研究成果は、理化学研究所のスーパーコンピュータ 「富岳」(注6)を利用して得られたものです(課題番号:hp220331、hp230124、hp240148)。
用語解説
注1 第一原理計算
原子核や電子の配置など、物質を構成する基本的な構成要素のみをインプットとし、量子力学に基づきその性質をコンピュータ上でシミュレーションする方法。実験では観測できない微視的な情報を得られるため、物性の理解に重要な役割を果たす。
注2 ワニエセンター
電子の局在基底として得られるワニエ関数の重心。電子の分布や局在の特性を反映しており、分子や固体の極性や電気的特性の計算に用いられる。
注3 ニューラルネットワーク
ある2つのデータの間の関数を多量のパラメータを持ったモデルで数値的に模倣する方法。データの組み合わせを予めサンプリングしておき、その組み合わせを再現するようにモデルを訓練させ、訓練されたモデルを未知のデータに適用することで予測を行う、という一連の流れを指す。
注4 分子動力学法
原子や分子の運動をコンピュータ上でシミュレーションする手法。物質の構造、物性、化学反応、相転移などの理解に重要な役割を果たし、特に分子液体や生体分子の研究に広く利用されている。
注5 現代分極理論
物質中の電気的な分極(双極子モーメント)の量子力学的な性質を記述する理論。ワニエ関数やベリー位相などの量子力学的概念を導入して電子の波動関数の変化として分極を捉える。
注6 スーパーコンピュータ「富岳」
スーパーコンピュータ「京」の後継機として理化学研究所が設置し、2021年3月から共用を開始した計算機。2020年6月以降、世界のスーパーコンピュータに関するランキングにおいて、4部門で4期連続1位、うち2部門で9期連続1位を獲得するなど、世界トップレベルの性能を持つ。


