
2023-11-02 米国国立再生可能エネルギー研究所(NREL)
◆米国再生可能エネルギー研究所(NREL)の研究者は、機械学習ツール「PolyID: Polymer Inverse Design」を使用して、分子構造に基づいて材料の特性を予測し、特定の用途に適したポリマーデザインの候補リストを作成することができるようにしました。
◆このツールは、持続可能で高性能なポリマーを見つけるプロセスを迅速かつ容易にする役割を果たし、食品包装、自動車部品、個人用保護具など、さまざまな用途に適したポリマーを探すために使用されています。
<関連情報>
- https://www.nrel.gov/news/program/2023/pick-your-polymer-properties-and-this-nrel-tool-predicts-how-to-achieve-them-with-biomass.html
- https://www.sciencedirect.com/science/article/pii/S2666389921002233?via%3Dihub
- https://pubs.acs.org/doi/10.1021/acssynbio.1c00189
- https://www.nature.com/articles/s41467-022-35237-x
グラフ・ニューラル・ネットワークを用いた既知および仮説の結晶のエネルギーと安定性の予測 Predicting energy and stability of known and hypothetical crystals using graph neural network
Shubham Pandey, Jiaxing Qu, Vladan Stevanović, Peter St. John, Prashun Gorai
Patterns Published: September 30, 2021
DOI:https://doi.org/10.1016/j.patter.2021.100361
")
Highlights
•A GNN is trained to predict total energy of ground-state and high-energy crystals
•The importance of a balanced training dataset is demonstrated
•The model ranks polymorphic structures of compounds with correct energy ordering
The bigger picture
Large-scale ab initio calculations combined with advances in structure prediction have been instrumental in inorganic functional materials discovery. Currently, only a small fraction of the vast chemical space of inorganic materials has been discovered. The need for accelerated exploration of uncharted chemical spaces is shared by experimental and computational researchers. However, structure prediction and evaluation of phase stability using ab initio methods is intractable to explore vast search spaces. Here, we demonstrate the importance of a balanced training dataset of ground-state (GS) and higher-energy structures to accurately predict their total energies using a generic graph neural network. We demonstrate that the model satisfactorily ranks the structures in the correct order of their energies for a given composition. Together, these capabilities allow the model to be used for fast prediction of GS structures and phase stability and for the facilitation of new materials discovery.
Summary
The discovery of new inorganic materials in unexplored chemical spaces necessitates calculating total energy quickly and with sufficient accuracy. Machine learning models that provide such a capability for both ground-state (GS) and higher-energy structures would be instrumental in accelerated screening. Here, we demonstrate the importance of a balanced training dataset of GS and higher-energy structures to accurately predict total energies using a generic graph neural network architecture. Using∼16,500 density functional theory calculations from the National Renewable Energy Laboratory (NREL) Materials Database and ∼11,000 calculations for hypothetical structures as our training database, we demonstrate that our model satisfactorily ranks the structures in the correct order of total energies for a given composition. Furthermore, we present a thorough error analysis to explain failure modes of the model, including both prediction outliers and occasional inconsistencies in the training data. By examining intermediate layers of the model, we analyze how the model represents learned structures and properties.
機械学習可能な13Cフラクソミクスのための計算フレームワーク Computational Framework for Machine-Learning-Enabled 13C Fluxomics
Chao Wu, Jianping Yu, Michael Guarnieri, and Wei Xiong
ACS Synthetic Biology Published:October 27, 2021
DOI:https://doi.org/10.1021/acssynbio.1c00189
Abstract

13C metabolic flux analysis (MFA) has emerged as a powerful tool for synthetic biology. This optimization-based approach suffers long computation time and unstable solutions depending on the initial guess. Here, we develop a machine-learning-based framework for 13C fluxomics. Specifically, training and test data sets are generated by metabolic network decomposition and flux sampling, in which flux ratios at metabolic nodes and simulated labeling patterns of metabolites are used as training targets and features, respectively. To improve prediction accuracy and simplify the model, automated processes are developed for flux ratio selection based on solvability and feature screening based on importance. We found that predictive performance can be significantly improved using both amino acids and central carbon metabolites in comparison with amino acids alone. Together with measured external fluxes, the predicted flux ratios determine the mass balance system, yielding global flux distributions. This approach is validated by flux estimation using both simulated and experimental data in comparison with canonical 13C MFA. The approach represents a reliable fluxomics method readily applicable to high-throughput metabolic phenotyping, which highlights the advances of intelligent learning algorithms in synthetic biology, specifically in the Test and Learn stage of the Design–Build–Test–Learn cycle.
耐熱性ポリ(エチレンテレフタレート)ヒドロラーゼの足場を自然の多様性から調達する Sourcing thermotolerant poly(ethylene terephthalate) hydrolase scaffolds from natural diversity
Erika Erickson,Japheth E. Gado,Luisana Avilán,Felicia Bratti,Richard K. Brizendine,Paul A. Cox,Raj Gill,Rosie Graham,Dong-Jin Kim,Gerhard König,William E. Michener,Saroj Poudel,Kelsey J. Ramirez,Thomas J. Shakespeare,Michael Zahn,Eric S. Boyd,Christina M. Payne,Jennifer L. DuBois,Andrew R. Pickford,Gregg T. Beckham & John E. McGeehan
Nature Communications Published:21 December 2022
DOI:https://doi.org/10.1038/s41467-022-35237-x
Abstract
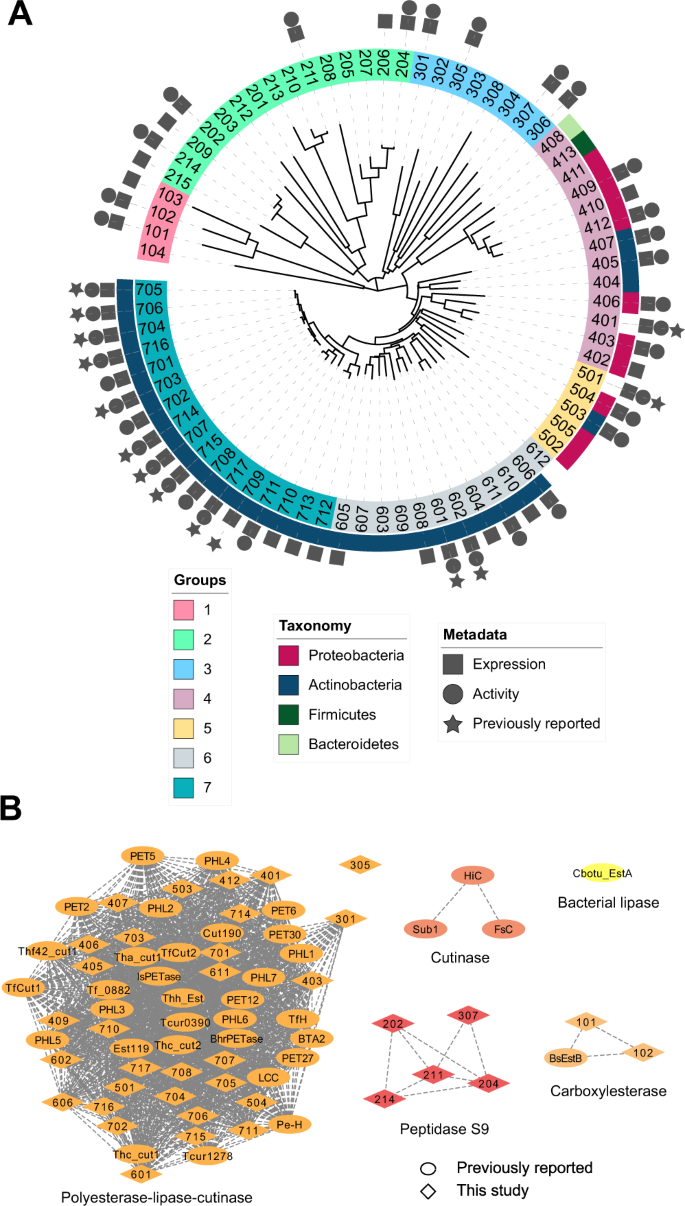
Enzymatic deconstruction of poly(ethylene terephthalate) (PET) is under intense investigation, given the ability of hydrolase enzymes to depolymerize PET to its constituent monomers near the polymer glass transition temperature. To date, reported PET hydrolases have been sourced from a relatively narrow sequence space. Here, we identify additional PET-active biocatalysts from natural diversity by using bioinformatics and machine learning to mine 74 putative thermotolerant PET hydrolases. We successfully express, purify, and assay 51 enzymes from seven distinct phylogenetic groups; observing PET hydrolysis activity on amorphous PET film from 37 enzymes in reactions spanning pH from 4.5–9.0 and temperatures from 30–70 °C. We conduct PET hydrolysis time-course reactions with the best-performing enzymes, where we observe differences in substrate selectivity as function of PET morphology. We employed X-ray crystallography and AlphaFold to examine the enzyme architectures of all 74 candidates, revealing protein folds and accessory domains not previously associated with PET deconstruction. Overall, this study expands the number and diversity of thermotolerant scaffolds for enzymatic PET deconstruction.

")
