時間のかかる理論計算を1万倍以上高速化し材料開発のプロセスを加速
2018-09-19 国立研究開発法人 産業技術総合研究所 ,国立大学法人 東京大学
ポイント
- 物理化学の知識と機械学習を合わせて、分子構造だけから分子の物性値を高速・高精度に予測
- 時間のかかる理論計算より1万倍以上速いため、材料開発のプロセスを大幅に加速可能
- これまで探索されてこなかった新たな物質の「発見」に繋がることを期待
概要
国立研究開発法人 産業技術総合研究所【理事長 中鉢 良治】(以下「産総研」という)人工知能研究センター【研究センター長 辻井 潤一】機械学習研究チーム 瀬々 潤 研究チーム長、椿 真史 産総研特別研究員は、 【総長 五神 真】 生産技術研究所【所長 岸 利治】(以下「東大生研」という) 物質・環境系部門 溝口 照康 准教授と共同で、化学物質の分子構造からその物質の物性値を高速、高精度に検証可能な形で予測する手法を開発した。
化学物質の物性値を知るには、実際に合成して物性値を測定するか、時間のかかる理論計算をする必要があった。今回、物理化学の知識に基づいて、分子中の原子間に、化学結合などの相互作用の「強さの変化」を「バネの伸び縮み」で表すような関数(ポテンシャル)を設定し、ニューラルネットで学習させた。この関数は、原子間の相互作用・化学結合の強さに対応するため、学習結果の物理化学的な解釈・検証が可能である。学習結果を用いて分子のさまざまな物性値を予測したところ、例えば原子化エネルギーについては100分の1秒の時間で誤差0.01 eV(電子ボルト)以下の精度で予測できた。これは、理論計算と同程度の精度を、理論計算の1万倍以上の速さで実現したことになる。
今回の成果の詳細はアメリカ化学会の物理化学分野の論文誌The Journal of Physical Chemistry Lettersに掲載されるが、それに先立ちJust Accepted Manuscriptが2018年8月6日に掲載された(DOI:10.1021/acs.jpclett.8b01837)。
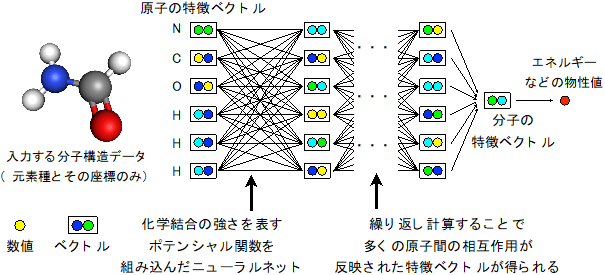
物理化学の知識を組み込んだ機械学習手法
開発の社会的背景
世の中には膨大な数の化学物質が存在する。アメリカ化学会の化学物質データベースには約3,000万種が登録されており、そのうち5,000種はそれぞれ世界で毎年1,000トン以上生産されている。これらの化学物質は、光の吸収や熱伝導などの強弱など、さまざまな物性値を示し、その物性値は、構成する元素の種類や分子の構造、化学結合の強さなどによって決まる。このような化学物質の物性値を知ることは材料開発において重要であり、これまで二つの方法が用いられている。一つは実際にその物質を合成しそれぞれの物性値を測定することと、もう一つは化学物質の分子構造をコンピューターに入力し理論計算することである。しかし、どちらもさまざまな設備と専門知識や経験が必要で、長い時間がかかる。これらの問題を解決し、化学物質が示すさまざまな物性値を高速・高精度で予測可能にすることで、材料開発のスピードを加速させる手法が求められている。
研究の経緯
産総研では、機械学習を、画像や言語などのデータのみならず、化学・材料データへ応用することを目指している。また、人間と相互理解できる次世代人工知能技術として、大規模なデータと人間が蓄積してきた知識をあわせて活用し、複雑な機械学習を可能にするとともに、学習の結果をより解釈・検証しやすくするための研究開発を進めている。今回、機械学習による化学物質の物性値予測に、物理化学の知識を利用する方法の研究開発に取り組んだ。
なお、今回の研究開発は、国立研究開発法人 新エネルギー・産業技術総合開発機構(NEDO)「次世代人工知能・ロボット中核技術開発/次世代人工知能技術分野/人間と相互理解できる次世代人工知能技術の研究開発」、国立研究開発法人 科学技術振興機構(JST) 戦略的創造研究推進事業 個人型研究(さきがけ)「理論・実験・計算科学とデータ科学が連携・融合した先進的マテリアルズインフォマティクスのための基盤技術の構築」研究領域(研究総括:常行 真司(東京大学 教授))における研究課題「情報科学手法を利用した界面の構造機能相関の解明」(研究者:溝口 照康(東大生研 准教授))の支援を受けて行った。
研究の内容
分子構造は、元素とその位置、つまり3次元立体構造データとして表現される。近年、どんな大きさや構造の分子でも、分子構造のデータをそのまま入力して物質の物性値を予測できる柔軟な機械学習の手法が提案され始めている。しかし、従来手法では学習結果の解釈が難しく、学習結果の妥当性が検証できないという課題があった。今回、物理化学の分野で用いられている近似式に基づいて、分子中の原子間に、化学結合などの相互作用の「強さの変化」を「バネの伸び縮み」で表すような関数(ポテンシャル)を設定した。そしてその関数を表すニューラルネット(機械学習モデル)を、大規模データで学習させる手法を考案した。この関数は、原子間の相互作用や化学結合の強さに対応するため、学習結果の物理化学的な解釈と検証が可能である。
今回考案した手法を用いて、13万を越える化合物からなる大規模なデータベースを学習させて、予測の計算時間と精度を評価した。その結果、分子が示すさまざまな物性値を理論計算の1万倍以上の速さで、理論計算と同程度の高精度で予測できることが確認できた(図1)。
また、原子間の化学結合(単結合や二重結合)の強さを示すポテンシャルについて、物理化学の理論計算によって得られたものと、今回の手法によって得られたものとを比較・検証したところ、それらが良く一致することが確認できた(図2)。これは、今回の手法が、物理化学的に解釈できる情報をデータから学習したことを示している。
物理学や化学などのデータについて何らかの予測を行う際には、コンピューターによる学習結果を、人間が既に持っている物理学や化学の知識や理論計算と照らし合わせて、その結果の妥当性を検証することが重要であるが、今回の手法は、物質の物性値の予測だけでなく、妥当性の検証も含めたプロセス全体を加速できる。
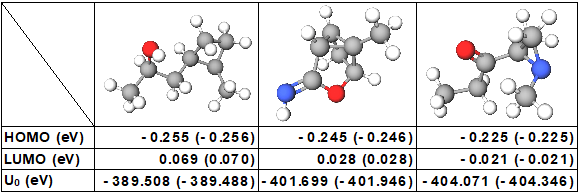
図1 機械学習で予測された物性値(HOMO、LUMO、U0)
括弧内は理論計算で得られた物性値
図中の原子はそれぞれ、白色が水素、灰色が炭素、赤色が酸素、青色が窒素である。
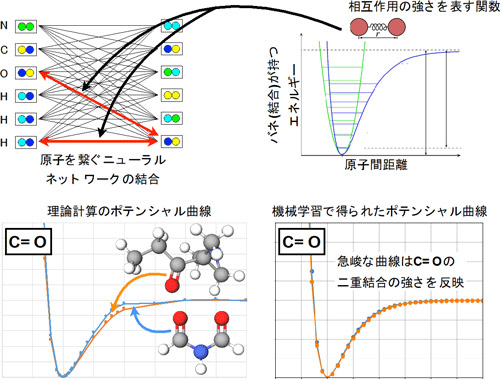
図2 化学結合の強さを示すポテンシャル曲線(左が理論計算、右が機械学習で得られた結果)
今後の予定
元素や分子構造の組み合わせの異なる化学物質は莫大にあるが、大量生産されているものは0.02 %に満たない。化学物質の物性値を高速・高精度で予測できる本手法によって多数の候補化学物質の物性値を網羅的に評価することで、より優れた機能や新しい機能を持つ化学物質を見出すプロセスを加速することが期待できる。今後は、今回開発した手法をより高精度化するとともに、手法を活用して材料開発を大幅に加速し、新たな化学物質の発見につなげていく。
用語の説明
- ◆理論計算
- 第一原理計算や格子静力学計算、分子動力学計算などさまざまな手法があり、ここでは原子の種類と配置から電子の波動関数を計算する第一原理計算を用いた。一回の理論計算には、数時間~数十時間の計算時間を要する。
- ◆機械学習
- データから変数間の関数関係や法則性を抽出し、データの分類や予測を可能にするための方法、アルゴリズム。人間の脳神経回路を模したニューラルネットワークや、サポートベクトルマシンなどさまざまな手法がある。
- ◆ポテンシャル
- 力の強さの変化を表す関数。ポテンシャル関数を微分したものが力の強さになる。
- ◆HOMO
- 最高被占軌道の略称。電子に占有されている最もエネルギーの高い分子軌道のことを指す。
- ◆LUMO
- 最低空軌道の略称。電子に占有されていない最もエネルギーの低い分子軌道のことを指す。
- ◆U0(原子化エネルギー)
- 原子化エネルギーとは、分子を構成する各原子になるまで引き離すときに、必要となるエネルギーのこと。